

Version AIONv3.20.0.2_IFUv57.2
These are the Instructions for Use (IFU) for AION variant interpretation platform manufactured by Nostos Genomics GmbH. These instructions provide general information and guidelines for use of the following product.
| Product identification | Version | Release date |
| AION | v3.20.0.2 | 28/07/2026 |

| Basic UDI | UDI-DI | UDI-PI |
| PP 11956 AION 62 | 13 11956 AION-V.3 0 47 | v.3.20.0.2-2026-07-28 |
AION can be accessed via Nostos Genomics Variant
Interpretation Platform 
The use of the device is subject to the term and conditions provided on the log-in screen, and to the ITA (Individual Tier Agreement).
ℹ️ If you want to request a demo, or have any doubts or questions, please get in touch with Nostos Genomics through:
| Manufacturer | Nostos Genomics GmbH |
| Address | c/o Stresemannstrasse 123 Tenant GmbH Stresemannstrasse 123 10963 Berlin Germany |
| Website | nostos-genomics.com |
| Assignment of the products to the registration numbers in the German Medical Devices Information and Database System |
Nostos Genomics GmbH is a CDS (clinical decision support) software manufacturer registered with ID DE/0000049631 and included at IFA Liste under ID 117315, and CA Registration ID: DE/CA73/238029-01. SRN DE-MF-000035385 |
Table 1: General information about the manufacturer
| Device ID | AION | |
| Basic UDI (BUDI) | PP 11956 AION 62 | |
| IVD/MD | IVD SaMD Clinical Decision Support (CDS) | |
| IVDR Class & Conformance Route | Rule 3-Class C-Genetic testing Annex IX-QMS Assurance | |
| Notified Body Name & ID# | BSI the Netherlands Say Building, John M. Keynesplein 9, 1066 EP Amsterdam, Netherlands Certification Body ID#: 2797 | |
| BfArM-Registration numbers | ID DE/0000049631 IFA Liste ID 117315 CA Registration ID: DE/CA73/238029-01 | |
| AION access (website) | https://app.nostos-genomics.com/ | |
| Product labelling |

|
|
Table 2: General information about the product
AION is an in vitro diagnostic medical device software intended to qualitatively process next generation sequencing (NGS) data from genetic testing. This AI-driven IVD software identifies genetic variants, predicts their classification based on the integration of multiple annotation sources and generates a variant ranking based on disease association. AION is a semi-automated clinical-decision support software to assist healthcare professionals in making informed medical decisions regarding clinically relevant genetic variants and genetic diseases.
AION’s indication of use relates to Mendelian genetic disease testing. Genetic testing involves the detection and assessment of specific alleles, variants and genotypes, that are associated with heritable traits and diseases or predispositions to disease for the individual or their descendants (diagnostic, screening and predictive testing).AION’s Intended patient population includes patients of all ages (prenatal, postnatal), sex, and ancestries who might undergo NGS sequencing analysis with the aim of testing for genetic diseases.
AION is intended to be used as part of the diagnostic workflow for NGS-based genetic testing by healthcare professionals (eg. human genetics professionals, geneticists, variant scientists, and healthcare practitioners with expertise in genetics). The software is not intended to be used by lay users.
The AION software is a cloud-based clinical decision support (CDS) tool designed to facilitate the interpretation of genetic variants in the context of genetic diseases. Its primary function is to predict the pathogenicity of genetic variants using artificial intelligence (AI) models, leveraging the interpretation of next-generation sequencing (NGS) data.
1. Input Data and Preprocessing:
ℹ️ Including HPO terms is crucial for improving variant ranking performance, particularly in small variants (singleton cases) and CNVs. For CNVs, the inclusion of HPO data is especially important, as it significantly enhances ranking performance. We strongly recommend users provide HPO terms when analyzing CNVs to achieve optimal results.
2. Variant Annotation:
AION uses a comprehensive variant annotation pipeline that enriches the raw genetic data with relevant contextual information. This includes:
The complete list of annotations is as follows:
3. Variant Classification:
Variant classification is provided following three approaches:
4. Output and Interpretation:
This comprehensive approach integrates bioinformatic data processing, AI-driven predictive modeling, and detailed annotation to assist healthcare professionals in making informed clinical decisions regarding genetic diseases.
⚠️ AION is a clinical-decision support IVD SW, as such no medically-relevant decisions should be made without first consulting the appropriate healthcare professional and/or information on disease effects, prevalence or others.
AION incorporates an Artificial Intelligence (AI) model to support the interpretation of diagnostic data. The AI analyses the input genetic data and provides suggested results based on patterns identified during its training. While the AI has been validated on clinically relevant data, its outputs may occasionally differ from expected findings. The AI is designed to assist and support the user, not to replace professional judgment. All AI-generated results must be reviewed and verified by the responsible healthcare professional before being used for clinical decision-making.
The AI model has been trained and validated on data relevant to the intended use. It is not intended to detect conditions outside this scope. To ensure safe and effective use, healthcare professionals should:
This ensures that the AI functions as a reliable decision-support tool while safeguarding the accuracy and integrity of the diagnostic process.
The AI model may be updated periodically in line with regulatory requirements and quality management procedures. Users will be informed of any changes that may affect device performance or instructions for use.
This section provides specific information on the AI model embedded within the AION analytical algorithm. It explains its role within the device, its intended use, operational principles, and how it supports variant interpretation in a clinical context. This information complements the general description of the device and is provided to ensure transparency, clarity, and safe use of the AI-enabled components by qualified healthcare professionals.
Within the AION clinical workflow, the analytical algorithm integrates an AI/ML component together with established computational methods to support the classification of genetic variants. This component processes structured variant-level data enriched with curated reference information and produces probabilistic classifications into clinically relevant categories. The resulting outputs are combined with other analytical evidence within the AION system to generate variant-level classifications and a prioritised list of candidate disease-causing variants with their associated clinical diagnoses.
The AI model has been trained and validated on curated datasets designed to be representative of the intended clinical population, following established principles of data quality, fairness and bias mitigation. This ensures that outputs remain relevant across subgroups within the intended patient population.
The AI model has been validated on clinically relevant data and is designed to provide reliable decision-support outputs. As with all diagnostic support tools, results should be interpreted in the context of other clinical and laboratory information. The AI complements, but does not replace, the professional judgment of qualified healthcare professionals. All outputs are assistive in nature and are to be reviewed and confirmed by the responsible healthcare professional before being used in clinical decision-making.
The deployed AI model is a locked version (non-adaptive in the field), ensuring that no further learning or modification occurs post-release. It is maintained under controlled configuration management in accordance with relevant software lifecycle standards, with traceability to the training data, preprocessing pipelines, and source code version. All algorithmic outputs are presented within the system’s user interface (UI) alongside supporting evidence and explanatory metrics to aid interpretation.
All outputs are assistive in nature and require review and interpretation by qualified healthcare professionals. The AI module, referred to as Circe and implemented as an integrated subcomponent within the analytical algorithm of AION, serves to support variant classification by processing annotated germline variants and generating probabilistic pathogenicity assessments. It leverages multiple sources of genomic and clinical evidence to provide calibrated predictions that contribute to the overall variant classification framework. Outputs are assistive in nature, form part of the AION analytical algorithm workflow, and require review and interpretation by qualified healthcare professionals.
The analytical algorithm component of AION is intended to identify diagnostic clinically relevant small variants (SNVs, indels), copy number variants and certain structural variants through automated variant interpretation (i.e. variant annotation, classification and prioritization), based on integrated molecular and phenotypic evidence. These outputs are intended to aid qualified healthcare professionals in diagnosing, screening or predicting the risk of genetic diseases in individuals or their descendants. The AI module within AION, is intended to assist in the classification of germline genetic variants identified through next generation sequencing (NGS) as disease-causing or benign in the context of Mendelian genetic disease testing.
This module is designed for use exclusively within the AION software environment and is not intended to be used as a standalone tool for direct medical decision-making.
The intended patient population includes individuals of all ages (prenatal and postnatal), sexes and ancestries, whose genomic data has been generated through validated NGS workflows for the purpose of genetic disease testing.
The intended users are licensed healthcare professionals (e.g., human genetics professionals, geneticists, laboratory professionals, variant scientists and healthcare practitioners with expertise in genetics). The software is not intended to be used by lay users or for self-testing.
No specialized training beyond the standard professional competence in genomic variant interpretation is required; however, users are expected to understand the principles of variant interpretation, and to have access to AION’s Instructions for Use (IFU). Users must also be aware that the analytical algorithm includes an AI/ML-based module for variant classification. The outputs are probabilistic in nature, reflect the operation of a trained statistical model, and must always be interpreted in context with other clinical and laboratory information
The analytical algorithm component operates within the AION system to facilitate the interpretation of genetic variants in the context of genetic diseases. Its primary function is to leverage available case-level genetic and clinical data to detect clinically relevant genetic variants. This component processes case-level genetic data (i.e. VCF files) and when available, phenotypic features described using Human Phenotype Ontology (HPO) terms. It does not ingest raw specimens or raw sequencing reads.
The algorithm applies validated computational methods, combining curated genetic knowledge, established clinical guidelines, and statistical, algorithmic and AI-based approaches, to classify variants and prioritise candidate diagnoses. The outputs consist of variant classifications, phenotypic similarity measures, and prioritised lists of candidate diagnoses, which are presented for review by qualified healthcare professionals.
The analytical algorithm component, including the Circe AI module, is fully embedded in the AION software ecosystem and cannot be deployed or operated as a standalone tool. Therefore, its intended operational environment is identical to that of AION. This includes use in professional healthcare or genetic testing laboratory environments, within the cloud-based AION infrastructure, accessed via supported web browsers and meeting the hardware/software requirements defined in the AION Instructions for Use (IFU).
The AI/ML subsystem embedded in AION operates within clearly defined boundaries of transparency, autonomy, human oversight, trustworthiness, and usability. Requirements are established to ensure that users understand the model’s operational limits, that misuse is anticipated and mitigated, and that outputs are communicated in a clinically interpretable way.
The AI module is a locked model. It does not adapt or learn in the field and therefore maintains consistent behaviour across all use cases within its validated scope. Transparency is supported through the presentation of outputs that can be traced back to the underlying evidence and decision logic. In the UI, variant classifications are accompanied by uncertainty indicators, supporting evidence references, and explanatory metrics to facilitate interpretation. Explainability is addressed through both design-time documentation and user-facing features, allowing healthcare professionals to understand the basis of model outputs.
All outputs from the AI module are intended as decision-support and must be reviewed by qualified professionals. AION is not an autonomous diagnostic system, and its outputs are not to be used as standalone diagnostic conclusions. The system design ensures that human operators remain in the loop, particularly in cases where model confidence is low or where clinical contexts extend beyond validated scope.
To further ensure reliability, users are expected to confirm AI-suggested results against their own expertise and patient-specific information. Any unexpected or inconsistent outputs should be reported through the established feedback and customer support channels, allowing continuous monitoring of device performance and safeguarding the accuracy of the diagnostic process.
Predictability and fairness are addressed through systematic validation, ensuring that outputs behave consistently across a range of representative datasets. Known limitations and residual risks, including contexts with reduced model confidence or unsupported variant classes, are explicitly communicated through warnings in this IFU and within the AION interface. Safety and misuse prevention are managed through technical safeguards, user training, and clear communication of operational boundaries. Misuse scenarios, such as application outside the intended patient population or reliance on outputs without professional oversight, are anticipated and mitigated through the intended use, warnings, interface design, and system restrictions.
AION as a software system may undergo periodic updates in line with regulatory requirements and quality management procedures. Such updates may include improvements to the user interface, additional non-AI features, security enhancements, or maintenance of system components. Users will be informed of any updates that affect device performance or these instructions for use.
The AI model itself is deployed as a locked version, meaning it does not adapt or learn during use in the clinical environment. In the event that a new locked model version is released, this update will follow the controlled regulatory processes. Users will be notified of any new validated performance metrics, together with any changes or limitations relevant for clinical interpretation.
All significant AI/ML-related residual risks are included in the general Warnings and Limitations section of this IFU. These risks are communicated transparently to the user, ensuring that the AI component is understood as one part of the broader AION analytical algorithm, with performance metrics aligned with those of the AION system as a whole, covering sensitivity, specificity, accuracy and reproducibility as defined in the device’s performance evaluation.
AION’s tertiary analysis approach based on AI/machine learning effectively automates the process of variant interpretation of genetic tests for the diagnosis of rare genetic disorders. Machine learning (ML) and automated tools on genetic variant classification, prioritization, and interpretation in the context of diagnosing rare diseases have significantly enhanced the accuracy, efficiency, and reliability of genetic diagnostics by integrating deep phenotyping, genotype-phenotype knowledge, and advanced computational methods (AION’s scientific validity).
AION’s clinical evidence is gathered from published experience from routine diagnostic testing supported by other sources of clinical performance data, as synthetic datasets that simulates the complexities and variability of genomic data.
AION’s performance is evaluated based on the state of the art benchmarking approach for NGS-based genetic testing. It addresses both analytical and clinical AION’s performance specifications per genetic variant type (small variants and CNVs/SV) from a retrospective real patient data submitted by diagnostic laboratories across Europe, encompassing individuals who underwent clinical exome and genome sequencing for diagnostics of rare genetic disease, being supplemented by other sources of data as synthetic cohorts from a GIAB-derived benchmark “truth set”. AION’s analytical performance is determined by the analytical sensitivity, confidence intervals and precision (repeatability/reproducibility). AION’s clinical performance is determined by the diagnostic sensitivity, confidence intervals, PPV, rank (position of the causative variant in the prioritized variant list) and total number of unique variants (total number of variants in the prioritizer list). Analytical and clinical performance are presented separately for the two prioritization lists provided by AION: AION Clues (AC) and AION Smoking Guns (SG); CNVs/SVs are reported under AC.
A summary of AION’s analytical and clinical performance results is shown in the subsection below.
A retrospective clinical cohort of 330 cases was assembled from real patient data submitted by diagnostic laboratories across Europe, encompassing individuals who underwent clinical exome and genome sequencing for diagnostics of rare genetic disease. These positive cases were carefully selected to evaluate AION because they reflect the diversity encountered in real-world diagnostic settings, including both technical complexities from varied laboratory workflows and clinical challenges presented by edge cases. Diagnostic variants were defined as variants considered clinically relevant and reported back to the clinician based on genetic, phenotypic, and segregation information from the patient.
Utilizing data from routine diagnostic testing offers significant value by providing insights from larger, heterogeneous and more complex populations. This cohort captures variations in wet lab protocols, sequencing approaches, secondary analysis workflows and the quality of clinical information available for analysis. Overall, the cohort included singletons and trios for small variants (trios exclusively in small variants), while CNVs/SVs were represented only as singletons. The cohort included 188 cases analyzed using the GRCh37 reference genome, including 135 cases with small variants and 53 cases with copy number variations (CNVs). The remaining 142 cases utilized the GRCh38 reference genome and included 106 cases with small variants and 36 cases with CNVs. For GRCh37, the cohort consisted of 40 trios (all with small variants) and 146 singletons (93 with small variants and 53 with CNVs). For GRCh38, the cohort included 79 trios (all with small variants) and 64 singletons (28 with small variants and 36 with CNVs)
To evaluate the influence of phenotypic data on variant interpretation, all cases were initially submitted with Human Phenotype Ontology (HPO) terms and subsequently re-analyzed without them. This diverse distribution of reference genomes, variant types and case structures ensures that the evaluation of AION’s performance accurately reflects the varied genomic configurations and clinical scenarios encountered in everyday diagnostic settings. This diversity is essential to identify less common, potentially serious device limitations and adverse events. By incorporating this comprehensive and varied data, our aim is to demonstrate AION’s capability to perform reliably across a wide range of clinical environments, ensuring robust and accurate variant interpretation in routine diagnostic workflows.
Table 3: Analytical and clinical performance results from the retrospective clinical cohort for SNVs (n=241) and CNVs/SVs (n=89). Analytical and Clinical performance is presented separately for the two prioritization lists provided by AION: AION Clues (AC) and AION Smoking Guns (SG). Results presented include a subset of the analytical performance results (analytical sensitivity, including confidence intervals), as well as clinical performance results (diagnostic sensitivity, including confidence intervals, positive predictive value (PPV), average variant rank and average number of unique variants). Analytical and clinical performance is presented separately for the two prioritization lists provided by AION: AION Clues (Table 3A) and AION Smoking Guns (Table 3B). CI: Confidence Interval, PPV: Positive Predictive Value.
| Analytical performance results | Clinical Performance results | |||||||
| Clinical cohort | Dataset | Analytical Sensitivity | Analytical Sensitivity CI (95%) | Diagnostic Sensitivity (Top‑3) | Diagnostic Sensitivity CI (95%) | PPV | Rank | Average # unique variants* |
| GRCh37 with HPOs | Small variants (n= 135) | 86.20% | 79.1%–91.6% | 80.10% | 72.29%–86.61% | 93.80% | 1.98 ± 0.03 | 9.51 ± 0.38 |
| CNVs (n= 53) | 92.40% | 81.8%–97.9% | 92.4% | 74.66% - 94.52% | 94.60% | 1.4 ± 0.15 | - | |
| GRCh37 without HPOs | Small variants (n= 135) | 86.20% | 79.1%–91.6% | 51.90% | 43.01%–60.72% | 90.70% | 5.07 ± 0.53 | 10.3 ± 0.4 |
| CNVs (n= 53) | 90.50% | 79.3%–96.8% | 90.5% | 79.3% - 96.8% | 94.50% | 1.8 ± 0.2 | - | |
| GRCh38 with HPOs | Small variants (n= 106) | 85.05% | 76.8%–91.2% | 70.10% | 60.48%–78.56% | 93.70% | 2.56 ± 0.3 | 9.3 ± 0.4 |
| CNVs (n= 36) | 86.10% | 70.5%–95.3% | 80.56% | 63.98% - 91.81% | 94.2% | 1.6 ± 0.2 | - | |
Table 3A. Retrospective clinical cohort — AION Clues
*For CNVs/SVs, the “Average # unique variants” is not reported. Unlike small variants, CNV/SV analysis typically yields only a very limited number of candidates per case, making this metric not meaningful in the context of CNV prioritization. Therefore, results for CNVs/SVs are presented in terms of sensitivity, PPV and rank only.
| Analytical performance results | Clinical Performance results | |||||||
| Clinical cohort | Dataset | Analytical Sensitivity | Analytical Sensitivity CI (95%) | Diagnostic Sensitivity (Top‑3) | Diagnostic Sensitivity CI (95%) | PPV | Rank | Average # unique variants |
| GRCh37 with HPOs | Small variants (n= 135) | 75.5% | 72.3%–86.6% | 70.3% | 61.6% - 77.9% | 93.8% | 1.3 ± 0.06 | 2.56 ± 0.18 |
| GRCh37 without HPOs | Small variants (n= 135) | 3.05% | 0.84%–7.63% | 1.5% | 0.19% - 5.41% | 36.7% | 1 ± 0.00 | 0.16 ± 0.4 |
| GRCh38 with HPOs | Small variants (n= 106) | 70.09% | 60.4%–78.5% | 61.6% | 51.7% - 70.9% | 93% | 1 .1 ± 0.03 | 1.68 ± 0.1 |
Table 3B. Retrospective clinical cohort — AION Smoking Guns
Note: Smoking Guns (SG) results are reported for small variants only. CNVs/SVs are reported exclusively under AION Clues (AC), as SG is not available for CNVs/SVs in AION.
The small variant analysis cohorts provide real-world evidence of AION’s use cases, reflecting its application in diverse clinical scenarios. For GRCh37, analytical sensitivity was 86.20%, while GRCh38 achieved a sensitivity of 85.05%. Diagnostic sensitivity was 80.10% for GRCh37 and 70.10% for GRCh38. These datasets include borderline and complex cases, offering valuable insights for performance optimization. Although the diagnostic sensitivity is slightly lower than that observed in synthetic datasets (see Summary of Performance Data from Other Sources available for comparative purposes), AION consistently prioritized disease-causing variants with an average rank of ~2 (GRCh37: 1.98 ± 0.03; GRCh38: 2.56 ± 0.3), demonstrating its reliability in handling diverse and challenging cases.
While HPO data do not directly affect AION’s analytical performance metrics, their inclusion significantly improves diagnostic sensitivity, confidence intervals, and variant ranking. Running AION without HPOs results in a substantial reduction in ranking performance, with the causative variant around the 5th position on average (5.07 ± 0.53) compared to ~2nd (1.98 ± 0.03) with HPOs. This underscores the importance of HPOs in enhancing AION’s diagnostic capabilities. Consistently with this, SG shows strong performance when HPOs are provided but degrades without phenotype input; AC remains robust for sensitivity and mainly benefits from HPOs through improved ranking and review efficiency.
These findings align with observations from synthetic cohorts, although the effects of HPO inclusion are more pronounced in real-world cases than in GIAB-derived synthetic datasets. This indicates that HPOs are especially critical for improving AION’s diagnostic performance in clinical settings. Consequently, we strongly recommend including HPOs in the analysis, as advised in the Instructions for Use, to maximize diagnostic accuracy.
The inclusion of HPOs had a significant impact on clinical performance metrics, notably diagnostic sensitivity, confidence intervals, and variant ranking. Without HPOs, AION’s ranking performance decreased significantly, with the causative variant at ~5.07 ± 0.53 on the prioritized list compared to ~1.98 ± 0.03 with HPOs. This pattern was more prominent in the clinical cohort than in synthetic datasets, further emphasizing the relevance of clinical data in enhancing AION’s diagnostic sensitivity. Despite the reduced performance in clinical cohorts compared to synthetic datasets, the observed trends are consistent, underscoring AION’s adaptability to real-world variability and its potential to improve diagnostic outcomes in complex clinical environments.
Performance metrics differ between reference genomes, with GRCh37/hg19 outperforming GRCh38/hg38 in analytical and diagnostic sensitivity and confidence intervals. These differences may stem from the smaller GRCh38/hg38 cohort size, reduced annotation maturity, and variability in real-world cases. Clinical cohorts show lower sensitivity and wider confidence intervals compared to synthetic datasets, which rely on highly curated GIAB data with spiked-in variants and much larger sample sizes (>1,000 vs. 106/135). Despite these disparities, consistent trends across datasets underscore the need for optimized annotations and larger real-world datasets to further enhance AION’s diagnostic accuracy and clinical utility across reference genomes. Overall, these findings reaffirm the robustness of AION’s clinical performance across reference genomes and emphasize the importance of selecting appropriate annotations to maintain diagnostic accuracy and consistency in clinical workflows.
Copy Number Variants (CNVs) present unique challenges compared to small variants, primarily due to their reliance on upstream processes (e.g., sequencing and secondary analysis) to distinguish true disease-causing variants from false positives caused by sequencing artifacts. These challenges are further compounded by the relative rarity of CNVs in clinical practice, as they account for only about 10% of disease-causing variants in monogenic disorders. This scarcity limits the availability of robust benchmarking datasets for evaluating CNV detection and interpretation.
The analytical sensitivity for CNVs is 92.4% (95% CI: 81.8%–97.9%) for GRCh37 and 86.1% (95% CI: 70.5%–95.3%) for GRCh38, comparable to that of small variants. However, the wider confidence intervals reflect the inherent variability and uncertainty in accurately interpreting larger structural changes. In the GRCh37 cohort for CNVs (n=16), AION demonstrated a diagnostic sensitivity of 92.4% (95% CI: 74.66%–94.52%) and a precision (PPV) of 94.6%, highlighting its ability to accurately prioritize causative variants. The average rank of causative variants was 1.4 ± 0.15, underscoring its reliable prioritization capabilities. In the GRCh38 cohort (n=36), analyzed using the current pipeline (v3.16.1.0), diagnostic sensitivity (Top-3) was 80.56% (95% CI: 63.98%–91.81%), PPV 94.2%, and average rank 1.6 ± 0.2, providing a benchmark for its performance in different reference genomes.
Establishing a comprehensive benchmarking dataset for CNVs remains particularly challenging. The rarity of these variants, coupled with variability in detection due to differing sequencing technologies, creates significant hurdles. Precisely identifying breakpoints is another complication, making validation efforts even more difficult. Despite these obstacles, ongoing improvements to CNV workflows and efforts to establish more reliable benchmarking standards are critical to enhancing AION’s performance in this area and bolstering its overall clinical utility. Although diagnostic sensitivity is largely comparable with and without HPOs in GRCh37 CNVs, including HPO terms improves ranking (e.g., ~1.4 with HPOs vs ~1.8 without), so we strongly recommend providing HPOs whenever available. Although diagnostic sensitivity is largely comparable with and without HPOs in GRCh37 CNVs, including HPO terms improves ranking (e.g., ~1.4 with HPOs vs ~1.8 without), so we strongly recommend providing HPOs whenever available.
Clinical performance for CNVs is strongly influenced by the inclusion of HPO data. Although the absence of HPO terms does not significantly affect diagnostic sensitivity, it has a notable impact on variant ranking, underscoring the importance of phenotypic data in CNV/SV analyses. While the optional nature of the HPO feature provides flexibility for users without phenotypic data, including HPO terms ensures more accurate and clinically relevant variant ranking. For optimal CNV/SV analysis, we strongly recommend incorporating HPO data (see the Limitations of Use and Contraindications section). Despite ranking impacts, AION maintains its ability to identify clinically significant variants, ensuring its diagnostic sensitivity aligns with its intended role as a clinical decision support tool.
Regarding the impact of the reference genome, an updated GRCh38 cohort (n=36) is now available and shows results analytically comparable within the same performance range. Differences, particularly in confidence intervals, likely stem from the limited sample size in current CNV customer cohorts. Notably, PPV data remain consistent, although rank remains slightly tighter with HPOs across builds.
To address current limitations in datasets for CNV analysis, we are actively expanding our cohorts to include a greater number of samples, which will further enhance benchmarking reliability and performance evaluation.
AION’s benchmarking results highlight its effectiveness in addressing diagnostic challenges, handling complex cases, and supporting clinical laboratories in managing their workflows. Its continuous validation process ensures alignment with current standards and regulatory requirements, contributing to its reliability as a tool for genetic diagnostics. These findings underscore AION’s potential to improve diagnostic workflows and deliver better outcomes for patients with rare genetic diseases.
Our results demonstrate the robust analytical and clinical performance of AION, highlighting its reliability, adaptability and impact in advancing genetic diagnostics. Analytical evaluations validate AION’s technical precision and consistency, while clinical assessments underscore its effectiveness in real-world diagnostic settings. The inclusion of phenotypic information through HPO terms further strengthens performance, particularly in CNV cases. In small variants, results are presented for both AION Clues (AC) and AION Smoking Guns (SG): AC remains robust for sensitivity and benefits from HPOs mainly through improved ranking, whereas SG shows strong performance when HPOs are provided and degrades without phenotype input.
The comparison between GRCh37 and GRCh38 reference genomes illustrates AION’s adaptability across different genomic assemblies, with advanced annotation resources in GRCh38 contributing to improved prioritization outcomes. With expanded real-world CNV cohorts (GRCh37 n=53; GRCh38 n=36) and updated analyses, AION demonstrates high sensitivity, stable PPV and favorable rank across builds; while the dependence on HPOs is less pronounced than in earlier assessments, including HPOs remains strongly recommended to optimize prioritization and streamline clinical review. Ongoing efforts to expand clinical datasets, particularly for GRCh38, will further enhance its validation and address existing gaps.
The results of these evaluations position AION as a state-of-the-art solution in genomic medicine. Its analytical and clinical performance showcases AION’s ability to deliver accurate, efficient and actionable insights for rare disease diagnostics. By bridging analytical precision with real-world applicability, AION sets a new standard for the integration of artificial intelligence in clinical genomics, reaffirming its value as a transformative tool for modern medicine.
Of note, limitations to these performance data include but are not limited to:
Results gathered in the tables above are representative of AION’s analytical and clinical performance and obtained from AION version v.3.16.1.0. Data updated on August 15th, 2025.
For more information see the white paper “Enhancing Rare Disease Diagnostics: Updated Performance Evaluation and Advancements of the AION AI-Driven Variant Interpretation Platform” available at the Nostos Genomic’s website Nostos Genomics - Latest News (nostos-genomics.com), under the section of Resources/Whitepapers
Concerns over data privacy and security significantly limit access to large and diverse patient genetic and clinical datasets essential for robust validation. While real patient samples provide the most accurate assessments, these limitations prompted us to simulate genetic data for 4299 individuals with rare genetic diseases. We achieved this by introducing disease-causative variants into high-quality control reference genomes and linking this genetic data to its associated clinical phenotype. The variants and clinical data used in this process were meticulously identified and curated from reputable peer-reviewed publications and through collaborations with scientific research groups. This approach enabled the creation of an extensive and heterogeneous reference dataset, allowing for a comprehensive evaluation of AION’s variant interpretation capabilities at scale while effectively circumventing the privacy and security constraints associated with accessing actual patient genetic data. Results are presented separately for AION Clues (AC) and AION Smoking Guns (SG).
A comprehensive range of genetic scenarios was simulated to mirror the diverse conditions found in real-world clinical laboratories, ensuring AION’s reliability across various genomic configurations and data availability. This included both trios and singletons to evaluate performance in familial and individual cases, the use of GRCh37 and GRCh38 reference genomes for compatibility across different assemblies and simulations with and without clinical data encoded as Human Phenotype Ontology (HPO) terms to assess effectiveness under varying levels of phenotypic information.
ℹ️ It is important to note that these results from synthetic cohorts are not intended to serve as clinical evidence demonstrating the clinical performance of AION. Rather, they are included as supplementary data that can provide additional information on AION’s performance in synthetic cohorts, complementing the results obtained from real patient data.
Table 4: Analytical and clinical performance results from the synthetic validation cohort for Single Nucleotide Variants (SNVs) (n=42994299). Cases were analyzed as trios versus singletons to evaluate the impact of including parental genetic data. The analyses for the GRCh37 cohort were repeated with and without HPO terms to assess the effect of incorporating phenotype data. Additionally, the same cases were analyzed using reference genomes GRCh37 and GRCh38 to compare the impact of different genome assemblies on clinical performance. Results presented include a subset of the analytical performance results (analytical sensitivity, including confidence intervals), as well as clinical performance results (diagnostic sensitivity, including confidence intervals, positive predictive value (PPV), average variant rank and average number of unique variants). Analytical and clinical performance is presented separately for the two prioritization lists provided by AION: AION Clues (Table 4A) and AION Smoking Guns (Table 4B). CI: Confidence Interval, PPV: Positive Predictive Value.
| Analytical performance results | Clinical Performance results | |||||||
| Validation cohort | Dataset (n=4299) | Analytical Sensitivity | Analytical Sensitivity CI (95%) | Diagnostic Sensitivity (Top‑3) | Diagnostic Sensitivity CI (95%) | PPV | Rank | Average # unique variants |
| GRCh37 with HPOs | Trios | 95.79% | 95.42%–96.60% | 95.70% | 95.15%–96.37% | 94.79% | 1.08 ± 0.005 | 2.02 ± 0.005 |
| Singletons | 91.25% | 90.30%–92.08% | 91.25% | 90.30%–92.08% | 94.50% | 1.02 ± 0.005 | 1.98 ± 0.005 | |
| GRCh37 without HPOs | Trios | 95.90% | 95.90%–96.50% | 95.90% | 95.20%–96.50% | 94.80% | 1.63 ± 0.01 | 3.8 ± 0.02 |
| Singletons | 91.42% | 90.50%–92.20% | 91.40% | 90.50%–92.20% | 94.50% | 1.51 ± 0.01 | 3.8 ± 0.03 | |
| GRCh38 with HPOs | Trios | 96.05% | 95.42% - 96.6% | 96.05% | 95.4% - 96.6% | 94.8% | 1.07 ± 0.002 | 4.04 ± 0.03 |
| Singletons | 91.56% | 90.68% - 92.3% | 91.56% | 90.68% - 92.3% | 94.6% | 1.02 ± 0.002 | 3.9 ± 0.02 | |
Table 4A. Validation cohort — AION Clues
| Analytical performance results | Clinical Performance results | |||||||
| Validation cohort | Dataset (n=4299) | Analytical Sensitivity | Analytical Sensitivity CI (95%) | Diagnostic Sensitivity (Top‑3) | Diagnostic Sensitivity CI (95%) | PPV | Rank | Average # unique variants |
| GRCh37 with HPOs | Trios | 90.84% | 90.18%–91.90% | 90.80% | 89.90%–91.60% | 94.50% | 1.02 ± 0.001 | 1.15 ± 0.02 |
| Singletons | 87.00% | 85.90%–88.00% | 87.00% | 85.90%–88.00% | 94.30% | 1.01 ± 0.001 | 1.11 ± 0.02 | |
| GRCh37 without HPOs | Trios | 1.35% | 1.03%–1.74% | 1.35% | 1.03%–1.74% | 20.4% | 1.00 ± 0.00 | 0.01 ± 0.00 |
| Singletons | 1.35% | 1.03%–1.74% | 1.35% | 1.03%–1.74% | 20.4% | 1.00 ± 0.00 | 0.01 ± 0.00 | |
| GRCh38 with HPOs | Trios | 91.07% | 90.18% - 91.9% | 91.07% | 90.68% - 92.3% | 94.5% | 1.07 ± 0.002 | 1.15 ± 0.003 |
| Singletons | 87.4% | 86.19% - 88.2% | 87.23% | 86.19% - 88.2% | 94.3% | 1.01 ± 0.003 | 1.13 ± 0.008 | |
Table 4B. Validation cohort — AION Smoking Guns
The trio analysis demonstrates higher analytical sensitivity and improved confidence intervals for sensitivity, indicating reduced bias in the trio-based approach. Regarding clinical performance, results for small variants reveal enhanced diagnostic sensitivity with the trio approach, reflected in less biased sensitivity confidence intervals. There are no significant differences in Positive Predictive Value (PPV) or variant ranking. Importantly, incorporating parental genetic background (trios) significantly impacts the total number of identified variants and associated performance metrics in variant classification and prioritization. Clinical performance data align with the analytical performance trends, particularly in sensitivity and confidence intervals. However, variant rankings remain consistent, suggesting that while variant discovery and performance metrics improve with trios, the prioritization process itself remains stable. These conclusions apply to both AC and SG
Clinical information input (e.g., HPO terms) does not significantly affect AION’s analytical specifications. For clinical performance, results from synthetic cohorts show that HPO data have minimal impact on diagnostic sensitivity in the trio approach, as parental information provides sufficient clinical context. Conversely, in the singleton approach, where only the proband file is analyzed, diagnostic sensitivity significantly decreases in the absence of HPO terms. Furthermore, in singletons without HPO data, variant ranking drops, with the causative variant averaging the third position in the prioritized list, compared to the first position when HPO terms are included. Therefore, optimal variant prioritization (ranking) requires patient clinical data (HPO terms) as input. In SG, the role of HPOs is critical: these evaluations are designed as a strict prioritization challenge in which phenotypic data are required for meaningful results. Without HPO input, performance drops to non-actionable levels, while with HPOs both GRCh37 and GRCh38 demonstrate strong and consistent performance (see Table 4B). This dependency is explicitly acknowledged as a limitation in the Instructions for Use, emphasizing the importance of providing phenotype data whenever available.
AION’s analytical performance is not significantly affected by the choice of reference genome, regardless of the genetic analysis approach (singleton vs. trios). The observed trend of enhanced analytical performance in trios compared to singletons (e.g., for AC with HPOs: GRCh37 trios 95.79% vs. singletons 91.25%; GRCh38 trios 96.05% vs. singletons 91.56%) remains consistent across reference genomes. A similar trio-over-singleton pattern is observed for SG with HPOs (GRCh37 90.84% vs. 87.00%; GRCh38 90.77% vs. 87.54%)) remains consistent across reference genomes. Clinical performance evaluation indicates some differences between reference genomes; however, these differences are within acceptable performance criteria and align with expected outcomes. These findings confirm the robustness of AION’s clinical performance across reference genomes, emphasizing the importance of selecting appropriate annotations to ensure diagnostic accuracy and consistency in clinical workflows
The typical use environment is an office or genetic testing laboratory.
AION runs on cloud computing.
Browser specifications to run AION are as follows. Minimum hardware requirements to run app UI, the minimal requirements would be:
AION’s indications of use relate to mendelian genetic diseases, consequently interpretation of variants that may not follow traditional mendelian patterns (such as somatic variants implicated in cancer or pharmacogenetics, PGx) is currently excluded from the indications of use (“limitations of use or contraindications”). Thus:
AION mitigates the risk of use for the diagnosis of the aforementioned conditions out-of-device’s intended use through different approaches, including the design of the user interface for case submission, as well as quality controls within the genetic analysis pipeline.
Additional control mitigating actions to prevent the risk of AION reporting variants out of its intended use, include a meticulous manual curation of AION’s datasets (gene-disease dictionaries), in order to ensure that only clinically relevant Mendelian diseases are retained and variants included on AION’s intended use reported. This process effectively excludes the majority of somatic cancer-related diseases (exclusion criteria):
Of note, although AION’s analysis results could potentially include variants associated with somatic mutations, variants with companion diagnostic indications, or other variants listed as limitations/contraindications the mechanisms reported herein efficiently mitigate the reporting of these variants and ensure that only variants included on AION’s intended use are reported.
Nevertheless, AION is contraindicated for somatic variants, T21 and PGx, and it is responsibility of the healthcare professional as intended user of AION to acknowledge this potential foreseeable device’s misused.
Predictions generated by the software are probabilistic outputs and must not be interpreted as definitive classifications of variant pathogenicity. In particular, predictions for loss-of-function variants reflect the distributional properties of the available data and should be considered as inputs into the broader decision-support workflow. Final interpretation of variant significance requires integration with downstream filtering, annotation, and expert review steps.
Additional limitations of use of AION are related to:
Of note, to avoid these limitations additional performance datasets from real world data is used for AION’s performance evaluation.
Additional limitations to the state-of-art benchmarking approach for NGS tertiary analysis performance evaluation are indicated at the end of the Performance characteristics section.
Effect of HPO terms on variant
prioritization: The inclusion of HPO terms is crucial
for optimizing variant prioritization in genetic analysis. This
is particularly relevant for both small variants (singleton
cases) and CNVs/SVs. For small variants, the addition of HPO
terms enhances the performance of variant ranking, improving the
algorithm’s ability to identify clinically relevant findings.
Although the absence of HPO terms does not compromise the
diagnostic sensitivity of the software, it impacts its ability
to effectively prioritize variants, potentially delaying or
complicating clinical interpretation.
In the case of CNVs, the importance of HPO data is even greater
due to the inherent complexity of analyzing these types of
variants. Without phenotypic information, the software’s
capability to prioritize relevant CNVs is diminished, increasing
the risk of overlooking significant findings or misinterpreting
results. To mitigate this, it is strongly recommended to include
HPO terms for CNV/SV analysis. The absence of phenotypic data is
a limitation and a potential risk that may impact the analysis’s
clinical utility. Including HPO terms is essential for achieving
accurate and clinically acceptable analysis results.
Similarly, the AION Smoking Guns prioritization strategy is
highly dependent on HPO data. These evaluations are designed as
a strict prioritization challenge in which phenotypic context is
required for optimal performance. Without HPO terms, the
algorithm defaults to reporting only a minimal set of variants
that might be considered clinically relevant in the absence of
phenotype data. This leads to a marked degradation in ranking,
with even “crystal-clear” pathogenic variants no longer
consistently appearing in the relevant range. With HPO terms
provided, however, Smoking Guns demonstrates robust and
clinically meaningful performance.
Input VCF files quality. The quality of the VCF files, influenced by the secondary analysis performed by users, is crucial for optimal results, please see AION requirements for more information on VCF input data and recommendations for secondary analysis best practices.
Sequencing technologies. AION’s quality control module includes tools to detect potential artifacts associated with specific sequencing technologies. We have also implemented a control list of artifacts (including information from over 200 VCF files and from scientific literature, Maffucci et al, 2018), which filters out variants commonly associated with sequencing errors across various platforms. In addition in the IVDB (Internal Variant Database) feature the software compute frequencies internal to each lab, and they could add specific filters, according to its potential sequencing artifacts.
Short-read platforms. AION has been designed and thoroughly validated with Next Generation Sequencing data from short-read platforms, mainly from Illumina technology./ Data from IonTorrent platforms and its evolutions are supported, but have produced many artifacts. Before using it, we recommend to AION’s users that the VCF data has been filtered for common artifacts from their setup.
Long-read platforms. Data from long read technologies has not been thoroughly tested. If you need to run samples with this type of data, we recommend you get in touch so we can quickly troubleshoot any potential issues and make sure the results have high quality.
AION is a AI/ML-based tertiary analysis software. ML-based automated variant interpretation faces challenges such as incomplete analysis due to specialized algorithms, poor input data quality, and performance variability across diverse populations due to biased variant databases. Furthermore, though the algorithm is designed to classify genetic variants with high accuracy, it is not infallible. Incorrect variant classification predictions by ML-based algorithms might lead to the exclusion of clinically relevant variants from analysis and results (false negatives or false positives).
As a clinical decision support software, we recommend the healthcare professional to use the algorithm’s results as a supportive tool rather than the sole basis for clinical decisions, and recommend that they conduct additional validation or consult with clinical experts when interpreting results to ensure accurate diagnosis and patient care.
Additional precautions or warnings are disclosed at AION’s Safety information section within this Instructions for Use.
Users can request or schedule training sessions during the demo and onboarding trial period. These sessions are designed to cover the initial setup of AION, the key functionalities of AION and learn how to use them effectively, and common troubleshooting and support strategies.
To schedule a session, please contact us at:
However, these Instructions for Use provide all the essential information needed for the safe and effective operation of AION. These instructions also include visual support with clickable flows to enhance user guidance.
Additionally, in the “Example Use Cases” section, we provide practical examples illustrating how AION can assist in identifying causative variants in patients. These cases include both clinical information as well as VCF files for users’ training.
For more information or additional assistance, please feel free to contact our support team.
Please see AION-IFU section Safety Information for product safety information in English and applicable EU members’ languages.
The Summary of Safety and Performance is available here.
AION is available also as an API product; submitting cases, monitoring analysis status, and retrieving results can be performed programmatically via the AION REST API. The graphical user interface (GUI) remains accessible for versions up to and including 3.19.x.
This section provides an overview of API usage for healthcare professionals and laboratory bioinformaticians who integrate AION into their diagnostic workflows. For full technical API documentation, please contact support@nostos-genomics.com.
ℹ️ Using AION via API does not change its intended use, clinical outputs, or performance characteristics. All variant classification, prioritisation, and annotation capabilities described in this IFU remain fully available via the API.
Access to the AION API requires authentication using OAuth2-based credentials. Your organisation will be issued API credentials (client ID and access token) during account setup. These credentials must be included in all API requests.
⚠️ AION uses long-lived access tokens for certain integrations. As with all credentials, treat your API token with the same care as a password. If you suspect a token has been compromised, contact Nostos Genomics support immediately for revocation.
A standard AION analysis via the API follows this sequence:
completed.The API accepts the same inputs as the previous UI-based workflow. See VCF Format for full requirements. In summary:
Analysis results are returned as downloadable files accessible via API endpoints once the case is complete. Key outputs include:
filter_tag column indicates any quality flags
applied to each variant — see Artifact and Quality
Filter Tags.The following table summarises the main differences between UI-based and API-based usage:
| Feature | UI | API |
|---|---|---|
| Case submission | Form-based upload | Programmatic via REST endpoints |
| Analysis monitoring | Dashboard with status indicators | Polling or webhook callbacks |
| Result review | Interactive variant cards | Downloadable TSV/XLSX files |
| Manual ACMG rule modification | In-UI toggle per criterion | Not available via API; ACMG scores reflect automated computation only |
| Report generation | PDF report builder in UI | Not available via API; reports must be generated by the integrating system |
| Variant labelling (AION DB) | In-UI classification labels | Available via dedicated API endpoints |
| Gene panels | Available | Not available |
| AION Internal Variant Database (IVDB) | Available | Not available |
ℹ️ Manual ACMG rule modifications (e.g. upgrading PS2 strength or manually applying PP4) are not available through the API in this release. All ACMG classifications returned via the API reflect the automated scoring described in ACMG/AMP Variant Classification. If manual rule modification is required for your workflow, contact support@nostos-genomics.com to discuss options.
The API supports running analyses against specific AION versions. This allows laboratories to maintain consistency across a study or audit period, or to validate results against a known version before upgrading. The version string can be specified at case submission. Contact Nostos Genomics support for information on which versions are available in your environment.
The security requirements and data handling practices described in Minimum IT Requirements apply equally to API usage. In addition:
The full API endpoint documentation — including endpoint specifications, request/response schemas, authentication details, and example integrations — is available at https://aiontibody.nostos-genomics.com/v1/docs. For questions or access issues, contact support@nostos-genomics.com.
Please see AION-IFU section Safety Information for product safety information in English and applicable EU members’ languages.
The Summary of Safety and Performance is available here.
AION is available also as an API product; submitting cases, monitoring analysis status, and retrieving results can be performed programmatically via the AION REST API. The graphical user interface (GUI) remains accessible for versions up to and including 3.19.x.
This section provides an overview of API usage for healthcare professionals and laboratory bioinformaticians who integrate AION into their diagnostic workflows. For full technical API documentation, please contact support@nostos-genomics.com.
ℹ️ Using AION via API does not change its intended use, clinical outputs, or performance characteristics. All variant classification, prioritisation, and annotation capabilities described in this IFU remain fully available via the API.
Access to the AION API requires authentication using OAuth2-based credentials. Your organisation will be issued API credentials (client ID and access token) during account setup. These credentials must be included in all API requests.
⚠️ AION uses long-lived access tokens for certain integrations. As with all credentials, treat your API token with the same care as a password. If you suspect a token has been compromised, contact Nostos Genomics support immediately for revocation.
A standard AION analysis via the API follows this sequence:
completed.The API accepts the same inputs as the previous UI-based workflow. See VCF Format for full requirements. In summary:
Analysis results are returned as downloadable files accessible via API endpoints once the case is complete. Key outputs include:
filter_tag column indicates any quality flags
applied to each variant — see Artifact and Quality
Filter Tags.The following table summarises the main differences between UI-based and API-based usage:
| Feature | UI | API |
|---|---|---|
| Case submission | Form-based upload | Programmatic via REST endpoints |
| Analysis monitoring | Dashboard with status indicators | Polling or webhook callbacks |
| Result review | Interactive variant cards | Downloadable TSV/XLSX files |
| Manual ACMG rule modification | In-UI toggle per criterion | Not available via API; ACMG scores reflect automated computation only |
| Report generation | PDF report builder in UI | Not available via API; reports must be generated by the integrating system |
| Variant labelling (AION DB) | In-UI classification labels | Available via dedicated API endpoints |
| Gene panels | Available | Not available |
| AION Internal Variant Database (IVDB) | Available | Not available |
ℹ️ Manual ACMG rule modifications (e.g. upgrading PS2 strength or manually applying PP4) are not available through the API in this release. All ACMG classifications returned via the API reflect the automated scoring described in ACMG/AMP Variant Classification. If manual rule modification is required for your workflow, contact support@nostos-genomics.com to discuss options.
The API supports running analyses against specific AION versions. This allows laboratories to maintain consistency across a study or audit period, or to validate results against a known version before upgrading. The version string can be specified at case submission. Contact Nostos Genomics support for information on which versions are available in your environment.
The security requirements and data handling practices described in Minimum IT Requirements apply equally to API usage. In addition:
The full API endpoint documentation — including endpoint specifications, request/response schemas, authentication details, and example integrations — is available at https://aiontibody.nostos-genomics.com/v1/docs. For questions or access issues, contact support@nostos-genomics.com.
Updates to the annotation sources are not logged here.
| Version | Regulation | Release date | Changes |
|---|---|---|---|
| 3.20.0.2 | CE-labeled (IVDR) | 28/July/2026 | Fixed a PVS1 (ACMG) scoring bug affecting frameshift, nonsense, and canonical splice variants. |
| 3.20.0.1 | CE-labeled (IVDR) | 25/June/2026 | Bug fixes for ACMG criteria (PVS1, BP1, BP3, BP4, PM5, PM2_supp) and gnomAD version consistency in explainability text. Minor improvements to AIONtibody. |
| 3.20.0.0 | CE-labeled (IVDR) | 18/May/2026 |
ACMG/AMP point-based classification system:
Updated ACMG/AMP variant classification engine following the
ClinGen points-based scoring framework. Each evidence criterion
is assigned a numerical score based on its strength and
direction; the sum determines the final classification. VUS
variants are sub-classified on a scale from “Ice Cold” to “Hot”.
See ACMG/AMP Variant
Classification. API usage: AION is now available via API; case submission, analysis retrieval, and result processing can be performed programmatically via the AION REST API. The GUI remains accessible for versions ≤ 3.19.x. Full endpoint documentation at https://aiontibody.nostos-genomics.com/v1/docs. See API Usage. Relevant Variants tab (CNVs): Dedicated IFU documentation added for the Relevant Variants tab, clarifying inclusion criteria and sorting logic. See Relevant Variants — CNVs. ClinVar Nostos conflict resolution: Documentation added explaining how AION’s ClinVar_Nostos field consolidates conflicting
ClinVar submissions into a single classification. See
ClinVar
Classification and Conflict
Resolution.Artifact/quality filter tags: Filter tag concepts documented, explaining their effect on variant prioritisation. See Artifact and Quality Filter Tags. Annotation data updates: ClinVar updated to 18/04/2026. Pangolin precomputed scores added for hg38 SNVs. Pfam domains updated (hg19 and hg38, 2025-09-26). REVEL v1.3 added. See Data Sources. PolyQ region definition: Documentation added describing how AION computes PolyQ regions from protein sequence. See Annotation Processing Notes. Bug fixes and stability improvements. |
| 3.19.0.1 | CE-labeled (IVDR) | 14/April/2026 |
Fixed an NMD bug affecting variants on the negative
strand. Activated the Cypress test covering annotation file download. |
| 3.19.0.0 | CE-labeled (IVDR) | 11/March/2026 |
Updated transcript annotations for hg38. Added support to run multiple versions of AION via the API, enabling version-specific analysis and improved workflow flexibility. Migrated the IFU manual to the new documentation system. Implemented various bug fixes and stability improvements. |
| 3.18.0.0 | CE-labeled (IVDR) | 16/February/2026 |
Data update, including manually curated gene to disease and
disease to phenotype dictionaries. Adjusting the processed data transfer between AION and Varvis. API security improvements to reduce the likelihood of account misuse, information disclosure and other security aspects. Bug fixes. |
| 3.17.0.5 | CE-labeled (IVDR) | 15/January/2026 |
Bug fixes. Minor maintenance activities. |
| 3.17.0.4 | CE-labeled (IVDR) | 17/December/2025 |
Improving dependency management. Bug fixes. |
| 3.17.0.3 | CE-labeled (IVDR) | 03/December/2025 | Bug fixes. |
| 3.17.0.2 | CE-labeled (IVDR) | 27/November/2025 | Bug fixes. |
| 3.17.0.1 | CE-labeled (IVDR) | 25/November/2025 | Bug fixes. |
| 3.17.0.0 | CE-labeled (IVDR) | 25/November/2025 |
Data sources update, including moving all sources to use Refseq
transcripts. Keeping Smoking guns on top of the AION ranking. Bug fixes. |
| 3.16.3.0 | CE-labeled (IVDR) | 6/November/2025 |
Introduction of gene-disease-moi confidence scores in API
output. Improvement of API integration debugging. Improve security and reliability to better control AION versioning and improve release robustness. |
| 3.16.2.0 | CE-labeled (IVDR) | 23/October/2025 |
Develop API output so that API users can filter variants
annotated by AION on relevant transcripts based on AION
shortlists and ACMG criteria. Improve the ranking for all variants outside of AION clues. This change doesn’t affect clinical performance. Improve benchmarking efficiency for faster development. This is an internal improvement. Minor bug fixes. |
| 3.16.1.3 | CE-labeled (IVDR) | 9/September/2025 | Minor bug fixes. |
| 3.16.1.2 | CE-labeled (IVDR) | 4/September/2025 | Minor bug fixes. |
| 3.16.1.1 | CE-labeled (IVDR) | 27/August/2025 | Minor bug fixes. |
| 3.16.1.0 | CE-labeled (IVDR) | 2/July/2025 | Minor bug fixes. |
| 3.16.0.0 | CE-labeled (IVDR) | 27/June/2025 |
Enhancement on the CNV ranking. Optimisation of the prioritized variants. Exclusion of upstream and downstream variants from the AION Smoking guns and AION Clues variant lists. Minor bug fixes. |
| 3.15.1.7 | CE-labeled (IVDR) | 7/May/2025 | Minor bug fix |
| 3.15.1.6 | CE-labeled (IVDR) | 28/April/2025 |
Updated labeling Minor bux fix Minor infrastructure maintenance |
| 3.15.1.5 | CE-labeled (IVDD) | 14/March/2025 | Minor bug fix |
| 3.15.1.4 | CE-labeled (IVDD) | 10/March/2025 |
Minor infrastructure maintenance Minor bug fix |
| 3.15.1.3 | CE-labeled (IVDD) | 27/February/2025 | Minor bug fixes |
| 3.15.1.2 | CE-labeled (IVDD) | 21/February/2025 | Minor bug fixes |
| 3.15.1.1 | CE-labeled (IVDD) | 15/January/2025 | Minor bug fix. |
| 3.15.1.0 | CE-labeled (IVDD) | 15/January/2025 |
Minor bug fixes. Coverage information update for non-coding genes Data Sources Update |
| 3.15.0.0 | CE-labeled (IVDD) | 19/December/2024 |
Coverage information tab in the UI Data Sources Update Case filtering by status Minor bug fixes. |
| 3.14.1.4 | CE-labeled (IVDD) | 16/December/2024 | Minor bug fixes. |
| 3.14.1.3 | CE-labeled (IVDD) | 12/December/2024 | Minor bug fixes. |
| 3.14.1.2 | CE-labeled (IVDD) | 10/December/2024 | Minor bug fixes. |
| 3.14.1.1 | CE-labeled (IVDD) | 03/December/2024 | Minor bug fixes. |
| 3.14.1.0 | CE-labeled (IVDD) | 29/November/2024 |
Gene coverage information in the UI for cases launched from
internal secondary analysis pipeline. Minor bug fixes. |
| 3.14.0.0 | CE-labeled (IVDD) | 26/November/2024 |
Introduction of AION Smoking guns and AION Clues in AION results
for small variants. gnomAD v4.1.0 used for variant priorization. Introduction of viewer only users. Inclusion of gene coverage information. Minor bug fixes. |
| 3.13.1.3 | CE-labeled (IVDD) | 1/October/2024 | Minor bug fix. |
| 3.13.1.2 | CE-labeled (IVDD) | 30/September/2024 | Minor bug fixes. |
| 3.13.1.1 | CE-labeled (IVDD) | 17/September/2024 |
Maintenance infrastructure improvement Minor bug fix. |
| 3.13.1.0 | CE-labeled (IVDD) | 5/September/2024 |
Minor bug fixes. Improvement on case search functionality. |
| 3.13.0.1 | CE-labeled (IVDD) | 12/August/2024 | Minor bug fix. |
| 3.13.0.0 | CE-labeled (IVDD) | 7/August/2024 |
Case search functionality CNV relevant variants optimization Minor bugs fixes |
| 3.12.3.1 | CE-labeled (IVDD) | 25/July/2024 | Minor bug fix. |
| 3.12.3.0 | CE-labeled (IVDD) | 25/July/2024 |
Data sources update: inclusion of gnomAD v4.1.0 Minor bugs fixes. |
| 3.12.2.2 | CE-labeled (IVDD) | 16/July/2024 | Minor bugs fixes. |
| 3.12.2.1 | CE-labeled (IVDD) | 11/July/2024 | Minor bugs fixes. |
| 3.12.2.0 | CE-labeled (IVDD) | 10/July/2024 | Minor infrastructure maintenance |
| 3.12.1.1 | CE-labeled (IVDD) | 25/June/2024 | Minor bug fix |
| 3.12.1.0 | CE-labeled (IVDD) | 24/June/2024 |
Activation/Inactivation of cases functionality Minor bugs fix |
| 3.12.0.2 | CE-labeled (IVDD) | 11/June/2024 | Minor infrastructure maintenance. |
| 3.12.0.1 | CE-labeled (IVDD) | 03/June/2024 | Minor bugs fixes. |
| 3.12.0.0 | CE-labeled (IVDD) | 30/May/2024 |
Annotation data update Minor bugs fixes. |
| 3.11.1.1 | CE-labeled (IVDD) | 16/May/2024 | Upgrade to support increased AION usage. |
| 3.11.1.0 | CE-labeled (IVDD) | 16/May/2024 |
Minor bugs fix in filtering and variant
classification. Ability to Refresh stats in the AION DB. |
| 3.11.0.2 | CE-labeled (IVDD) | 8/May/2024 | Minor bug fix in gene to disease association. |
| 3.11.0.1 | CE-labeled (IVDD) | 2/May/2024 | Minor bug fix relating authentication issues in integrated services |
| 3.11.0.0 | CE-labeled (IVDD) | 25/April/2024 |
Data update for ClinVar and CNV annotations Link to IGV viewer application added Minor bugs fixes |
| 3.10.0.3 | CE-labeled (IVDD) | 10/April/2024 |
Improvement on information display in the user interface Bug fix on Advanced Filtering results filtering and display |
| 3.10.0.2 | CE-labeled (IVDD) | 08/April/2024 | Bug fix in showing status of the affectedness of parents |
| 3.10.0.1 | CE-labeled (IVDD) | 04/April/2024 | Bug fixes and new CNV file format support |
| 3.10.0.0 | CE-labeled (IVDD) | 26/March/2024 |
Ability to manually classifiy small variants and store comments
for each variant classification Ability to store small variants manual variant classification and VCF variants in an internal AION DB from every case, and use this variant information in new cases. |
| 3.9.6.0 | CE-labeled (IVDD) | 3/March/2024 | Silent release on functionality to support segregation/trio analyiss for CNVs pipeline |
| 3.9.5.1 | CE-labeled (IVDD) | 21/February/2024 | Fix on Manual Classification tabs display |
| 3.9.5.0 | CE-labeled (IVDD) | 21/February/2024 |
Support to combined vcf files (SNV and CNV) Small fixes and improvements for Layout for small variant cards Case status Advanced filters |
| 3.9.4.2 | CE-labeled (IVDD) | 13/February/2024 | Fix in report downloading |
| 3.9.4.1 | CE-labeled (IVDD) | 9/February/2024 | Fix in report downloading |
| 3.9.4.0 | CE-labeled (IVDD) | 8/February/2024 |
Filtering by disease inheritance and patient-disease phenotype
match available on both SNV & CNV table views through
advanced filters. UX improvement for filtering IVDB based filtering by introducing column level filters on the numeric data from IVDB Small fix on long gene list input in advanced filters |
| 3.9.3.1 | CE-labeled (IVDD) | 24/January/2024 |
Fix of issues related filtering by Empty through advanced
filters Dictionary fix to always keep the highest confidence for g2d association from a source Small fixes to advanced filtering and long username overflow issue |
| 3.9.3.0 | CE-labeled (IVDD) | 15/January/2024 |
Introducing filtering based on IVDB data in the advanced
filtering Visualisation of source and confidence information related the gene-disease-MOI associations in the UI Usability enhancements and fixes to advanced filtering functionality |
| 3.9.2.1 | CE-labeled (IVDD) | 5/January/2024 | Fixing issue related concurrent CNV runs |
| 3.9.2.0 | CE-labeled (IVDD) | 20/December/2023 |
Introducing advanced filtering in AION. Build filter queries,
save them into a reusable filter and apply quickly to all your
cases across the platform. Advanced filters are available on
both SNV and CNV manual filtering view and users can create
their own private filters or share them with the whole
organisation. Quality hard filters are now removed from the SNV annotation process, so all variants of the analysed regions where FILTER=PASS or “.” , regardless of quality ( DP, VAF ) will get annotated and are available on SNV manual filtering view for analysis. The filterable values are now static on SNV and CNV manual filtering column filters. |
| 3.9.1.0 | CE-labeled (IVDD) | 13/December/2023 | Minor improvements and bug fixes in the pipeline |
| 3.9.0.0 | CE-labeled (IVDD) | 29/November/2023 |
New version of the dictionaries used for gene to disease to mode
of inheritance associations. Minor bug fixes in the UI Support for CNV vcf files with only one field in the FORMAT column |
| 3.8.5.1 | CE-labeled (IVDD) | 21/November/2023 | Fix of bug affecting segregation information in subset of trio cases with an affected parent |
| 3.8.5.0 | CE-labeled (IVDD) | 16/November/2023 |
New design in the final report Support for CNVpytor VCF file format as well as DragenCNV with 0 POS |
| 3.8.4.1 | CE-labeled (IVDD) | 6/November/2023 |
CNV pipeline gnomAD SV threshold for relevant tab filtering
corrected to 0.01 Hubspot chatbot added Updated transcript file for hg19 to ensure always showing the latest version Small UI improvements |
| 3.8.4.0 | CE-labeled (IVDD) | 31/October/2023 |
New canonical transcript files Bug fix to gnomAD SV hg38 data liftover Small UI fixes |
| 3.8.3.1 | CE-labeled (IVDD) | 23/October/2023 |
Bug fix to long indel visualisation on AION ranking
view Infrastructure change on storing shared between components ACMG calculator code |
| 3.8.3.0 | CE-labeled (IVDD) | 17/October/2023 |
More information added to the UI from gnomAD Added possibility to generate report in Spanish language Bug fix to correct uncontinuos ranking |
| 3.8.2.0 | CE-labeled (IVDD) | 9/October/2023 |
Updated Global frequent artifact blacklist for hg38
cases Update to the PVS1 gene file and annotation strategy Update to handling of high frequent whitelisted variants IVDB refresh endpoint memory issue fix |
| 3.8.1.1 | CE-labeled (IVDD) | 3/October/2023 | Fix to the case history version numbering schema |
| 3.8.1.0 | CE-labeled (IVDD) | 2/October/2023 |
Introducing ability to edit patient phenotype directly from the
case page and recalculate results Internal variant database data loading related bug fixes and improvements |
| 3.8.0.1 | CE-labeled (IVDD) | 20/September/2023 |
Fixed style of AION ranking tooltip Removed unavailable file to download for CNV-only cases Re-added manual link missing to case submit page |
| 3.8.0.0 | CE-labeled (IVDD) | 19/September/2023 |
Native annotation support in GRCh38 for small variants,
including gnomAD 3.1 and MANE Select transcript CNVs can now be analysed in GRCh38 as well Support for more CNV input formats Changes on submit case flow and submit case form allowing the possibility to submit CNVs only |
| 3.7.1.0 | CE-labeled (IVDD) | 4/September/2023 |
High quality variants in CNV table is now called relevant
variants and combines filtering criteria for quality + gnomAD SV
allele frequency of known cnvs overlapping UI fixes to CNV view |
| 3.7.0.1 | CE-labeled (IVDD) | 21/August/2023 |
Small UI fixes to CNV manual filtering view Bug fix for symptoms comparison dialog on CNV drawer |
| 3.7.0.0 | CE-labeled (IVDD) | 14/August/2023 |
UI and manual filtering functionality for CNV results CNV results are now visualised and available for manual filtering in a new CNV manual filtering view. |
| 3.6.0.4 | CE-labeled (IVDD) | 20/July/2023 | Bug fix to correct jointcalling for multiallelic variants and liftover |
| 3.6.0.3 | CE-labeled (IVDD) | 19/July/2023 | Bug fix to normalisation step |
| 3.6.0.2 | CE-labeled (IVDD) | 6/July/2023 | Update of gene panels available by default |
| 3.6.0.1 | CE-labeled (IVDD) | 30/June/2023 | AWS batch scaling optimisation |
| 3.6.0.0 | CE-labeled (IVDD) | 22/June/2023 |
New version of gene-disease-phenotype dictionaries New version of artifact denylist Small improvements to pipeline and infrastructure |
| 3.5.0.4 | CE-labeled (IVDD) | 9/June/2023 | FE bug fixes |
| 3.5.0.3 | CE-labeled (IVDD) | 8/June/2023 | FE bug fixes |
| 3.5.0.2 | CE-labeled (IVDD) | 7/June/2023 |
Improved error messaging from pipeline Bug fixes. |
| 3.5.0.1 | CE-labeled (IVDD) | 31/May/2023 |
CNV output formatting, pipeline bug fixes FE bug fixes. |
| 3.5.0.0 | CE-labeled (IVDD) | 25/May/2023 | Users can now run AION without HPO terms assigned to the case. We strongly suggest to include them to keep high ranking specificity |
| 3.4.0.0 | CE-labeled (IVDD) | 23/May/2023 |
AION now annotates CNV/SV variants provided with VCF files and
prioritises them with ACMG criteria. See more details in the
VCF
format UI improvements on case page: Supporting input file review and download Introducing fixed case header for better contextual information Archive functionality is now called delete to avoid confusions Enabling internal variant database statistics in manual filtering view |
| 3.3.1.0 | CE-labeled (IVDD) | 17/May/2023 | Implemented snackbar and maintenance screen to inform users of releases and maintenance periods |
| 3.3.0.0 | CE-labeled (IVDD) | 27/April/2023 |
Improved annotation infrastructure Improved gnomAD frequency annotation considering genome reference versions. Improved NMD, functional domain, Clinvar hotspot as well as splicing annotation Update of Clinvar gene lists (PVS1, PP2, BP1) Improvement of ACMG criteria (PS1, PM5, PM1, BP3) |
| 3.2.4.0 | CE-labeled (IVDD) | 12/April/2023 | Customer-specific internal variant database for small variants (SNPs and Indels) |
| 3.2.3.0 | CE-labeled (IVDD) | 16/March/2023 |
Reference genome of input files is asked when submitting a case.
The user input is verified with the data in VCF and the user is
informed about conflicts Manual filtering view now informs of number of variants passing the search & filter criteria Small FE improvements |
| 3.2.2.0 | CE-labeled (IVDD) | 9/March/2023 |
Update of VEP version and annotation infrastructure Infinite scrolling on manual filtering view to improve UX Ability to archive cases Small FE improvements Update of annotation pipeline includes running VEP on the fly. The VEP version used is 108. |
| 3.2.1.1 | CE-labeled (IVDD) | 20/Feb/2023 |
Adding ZenDesk for customer support Small FE improvements |
| 3.2.1.0 | CE-labeled (IVDD) | 6/Feb/2023 |
Update to ClinVar (21/01/2023) and disease dictionaries
(01/2023) Improvements to ACMG and ClinVar classification implementation Small FE fixes Updated ClinVar to newest release and updated internal ClinVar truth Updated ACMG Benign rules (BA1, BS1, BS2) Updated ACMG Pathogenic rules (PM4) Update whitelist of high frequently variants from full genes to variants Update variant filters Update of databases relating gene, disease and phenotype dictionaries Improvement of cosegregation consideration |
| 3.2.0.4 | CE-labeled (IVDD) | 1/Feb/2023 |
Bug fixed concerning reporting for old cases Terminology changes on login screen |
| 3.2.0.3 | CE-labeled (IVDD) | 24/Jan/2023 | Bug fixed concerning running cases with only few variants with partial annotations. |
| 3.2.0.2 | CE-labeled (IVDD) | 23/Jan/2023 |
Filtering on a column in the manual variant filtering view will
only show options from the available current column
values When sorting and filtering, sorting tags show “Sort by:” and appear first Removed the ability to add HPO terms to parents to avoid confusion. Fixed a bug preventing correct sorting by position Removed Intercom plugin Fixed error preventing tag in gene filter header from changing to “Applied” only when the filters were applied Fixed error when filtering of ClinVar column by ‘Conflicting interpretation’ Fixed message if no variants were found in the AION ranking Fixed issue showing negative GQ values caused by integer overflow Fixed issue causing non-unique HPO terms for cases created through API (didn’t affect users) Fix workflow and script for rd-aion-wrapper deployment through github actions Fixed minor bug in reading ‘summary’ Fixed minor bug in reading ‘case_info’ Installed Hotjar Improved annotation pipeline with refactorings Improved system usage Added relaunch case API to facilitate benchmarking and testing |
| 3.2.0.1 | CE-labeled (IVDD) | 18/Jan/2023 | Bug fixed concerning variants not being added to the report from the manual variant filtering view. |
| 3.2.0.0 | CE-labeled (IVDD) | 16/Jan/2023 |
Added manual filtering view for browsing and reporting annotated
variants In silico panels feature allowing restricting the analysed regions when submitting the AION analysis. Similar in silico panel and gene filters are available also on the manual filtering view for subsetting the annotated variants Improved consideration of affectedness of parents in trios and implementation of ACMG criteria related to cosegregation (PP1, BS4) AION pathogenicity classification calibrated into the following classes: Benign, Likely Benign, VUS, Likely pathogenic, Pathogenic Improved deny list of frequently seen artefacts to reduce number of prioritised artefacts Added patch version to AION version Investigation: fix CH logic for AR diseases with clear CH pathogenic variants Manual processing should error/warn of obsolete HPOs If vcf file is empty after downsampling, print proper error/warning Support analysing vcf with 1 only variant after downsampling Fix cosegregation_rank for CH without partner HPO in inheritance column for customer sample Implement segregation part for ACMG - PP1 Implement segregation part for ACMG - BS4 New filter: variants with MOI = AD and segregation = maternal or paternal Add CH fishing logic for Tier Null variants Skip PS2 for non joint called trios Include patient HPO terms in prioritizer file Curation of disease entries in our catalogue containing “susceptibility”, “resistance”, or being associated to expansions strategy to include/exclude related diseases Change message if no variants were found in the AION ranking Add BS2 BA1 as mutually exclusive |
| 3.1.0 | CE-labeled (IVDD) | 7/Nov/2022 |
Updated UI for case table, submit process, case details and
variant cards Updated ACMG implementation: Implementation of PS2, PVS1, PM2, PM3, PM6 The user can now apply/remove criteria as they see fit and the ACMG prediction is dynamically updated Criteria applied automatically by the AI can also be overruled and are marked with a symbol for explicability ACMG criteria grouped by segregation, disorder, effect, computational and functional, gene and region, frequency, other databases Wrong GTs for deletions after VT decomposition Prioritised file contains variants with filter tags AD read incorrect for multiallelic variants Create downsampling BED files with chr field Identify Tier CH for splice variants Fix sex chromosome segregation edge case Adapt vt normalize and remove any other chromosome not part of autosomal and sexual Support for VCF files not having AD but RO, AO fields Improve Compound heterozygous reporting Simplify variant status summary from Clinvar Add new link columns for google scholar and pubmed Get pipeline progress to reflect actual status Modify identification strategy for missing compound heterozygous partners Investigate de novo calling improvements Modularize ACMG calculation Define and implement new annotation file Missing RefSeq Transcript IDs (RefSeq_TranscriptID) Review and fix genes with wrong MOI from sources Include more genes in the white list Fix unsupported MOIs in current dictionaries Relax benign_combo filter Add manually curated gene to disease associations to dictionary Manual curation of MOI for mendelian genes missing MOI info Relax non_pathogenic filter criteria Define more relaxed filters for compound heterozygous variants Remove INFO field from VCF files (before vcf integrity) Include filter-reasoning column in additional unfiltered prioritizer output file Automatically filter unsupported regions to reduce the size of VCF Automatically liftover grch38 files to grch37 Define and set a default value for MIN_VAF Update gene panel for mendelian diseases Code refactoring of ACMG to on the fly in aion, integrate with latest AION dev Improve tiering gnomAD hard filters - extended solution Unify column name for ML score (nostos_interpretation, circe) ACMG: uniformize values in output files Include origin from ClinVar (for ACMG - PM6) Review and extend PVS1 & PM6 Review use of gnomAD_hom and gnomAD_AC to deprioritize variants Implement ACMG - PM2 Implement ACMG - PM6 Update Omim to disease dictionary Show intermediate HPO terms that match the patient Implement segregation part for ACMG - PM3 Implement segregation part for ACMG - PS Support more VCF formats Add version info to output files Improved normalization of VCF files Implement error/return codes into pipeline logging outputs Add more data to quality report Error coding schema for quality report Remove sorting from join calling Refactoring filtering logic Documentation of the blacklist file Standardize dev environment using docker-compose Integrate script for plots in benchmarking process Generate VCF quality report VAF filter (High-ranking variants in recurring genes (SON, KMT2C…): set quality filter at 20% VAF) Show intermediate HPO terms (ancestors) |
| 3.0.1 | CE-labeled (IVDD) | 1/Jun/2022 | Improvements on the genome build check |
| 3.0.0 | CE-labeled (IVDD) | 25/May/2022 |
CE-IVDR certified version Modularization and dockerization of AION pipeline Tiering of pedigree cases Improved scoring and management of compound heterozygous variants Links to external resources added: RefSeq, Ensemble, Transcripts Quality module introduced ensuring normalisation as well as handled errors for VCF format Bugs fixes to increase |
See safety information in different languages below: English, German, Swedish, Spanish, Italian
AION is an in vitro diagnostic medical device software intended to qualitatively process next generation sequencing (NGS) data from genetic testing. This AI-driven IVD software identifies genetic variants, predicts their classification based on the integration of multiple annotation sources and generates a variant ranking based on disease association. AION is a semi-automated clinical-decision support software to assist healthcare professionals in making informed medical decisions regarding clinically relevant genetic variants and genetic diseases.
Currently AION’s indication for use relates to mendelian genetic diseases testing. Consequently, interpretation of variants that may not follow traditional mendelian patterns is currently excluded from the indications of use. Thus:
See section of “Limitations of use and contraindications” for further information on these limitations as well as in the actions taken to mitigate potential risks of reporting this type of variants.
Using AION out of its current indications for use might lead to software malfunctioning and analysis failure; it would be done under sole responsibility of the user.
AION input data are VCF files. It is the responsibility of the user to provide high quality input VCF data to allow for optimal device output results. See for recommendations VCF format section.
AION supports NGS short reads technology on both Illumina and ION torrent, with the recommendations indicated as per Variant type, genomic regions and sequencing technologies. We recommend the users to contact our support team if using other types of NGS platforms.
⚠️ Any serious incident that has occurred in relation to the device shall be reported to Nostos Genomics GmbH (see contact details below) and to the competent authority of the EU Member State in which the user and/or the patient is established.
🗒️ For further information on safety and risks of the device, please contact us
Our website: Nostos
Genomics – AI-driven genetic analysis platform 
Our email: regulatory@nostos-genomics.com
AION ist eine Software für In-vitro-Diagnostika zur qualitativen Verarbeitung von Next Generation Sequencing (NGS)-Daten aus Gentests. Diese KI-gesteuerte IVD-Software identifiziert genetische Varianten, prognostiziert ihre Klassifizierung auf der Grundlage der Integration mehrerer Annotationsquellen und erstellt ein Varianten-Ranking auf der Grundlage der Krankheitsassoziation. AION ist eine halbautomatische Software zur Unterstützung klinischer Entscheidungen, die medizinisches Fachpersonal dabei unterstützt, fundierte medizinische Entscheidungen über klinisch relevante genetische Varianten und genetische Krankheiten zu treffen.
Gegenwärtig ist AION für die Untersuchung von genetischen Krankheiten im Mendelschen System vorgesehen. Folglich ist die Interpretation von Varianten, die nicht den traditionellen mendelschen Mustern folgen, derzeit von der Anwendungsindikation ausgeschlossen. Daher:
Die Verwendung von AION außerhalb seiner derzeitigen Anwendungsgebiete kann zu unerwartetem Softwareverhalten und Analysefehlern führen; dies geschieht auf alleinige Verantwortung des Benutzers.
AION-Eingabedaten sind VCF-Dateien. Es liegt in der Verantwortung des Benutzers, qualitativ hochwertige Eingabedaten bereitzustellen, um optimale Ausgabeergebnisse des Geräts zu ermöglichen. Siehe Abschnitt Empfehlungen für das VCF-Format.
AION unterstützt die NGS Short Reads Technologie sowohl von Illumina als auch von ION torrent, wobei die Empfehlungen je nach Variantentyp, genomischen Regionen und Sequenzierungstechnologien angegeben sind. Wir empfehlen den Nutzern, unser Support-Team zu kontaktieren, wenn sie andere Arten von NGS-Plattformen verwenden.
⚠️ Jeder schwerwiegende Vorfall, der im Zusammenhang mit dem Gerät aufgetreten ist, muss der Nostos Genomics GmbH (siehe Kontaktangaben unten) und der zuständigen Behörde des EU-Mitgliedstaates, in dem der Anwender und/oder der Patient ansässig ist, gemeldet werden.
🗒️ Für weitere Informationen zur Sicherheit und den Risiken des Produkts kontaktieren Sie uns bitte
Unsere Website: Nostos
Genomics – AI-driven genetic analysis platform
#safety-information
Unsere E-Mail: regulatory@nostos-genomics.com
AION är en medicinteknisk programvara för in vitro-diagnostik avsedd för kvalitativ bearbetning av NGS-data (Next Generation Sequencing) från genetiska tester. Denna AI-drivna IVDR-mjukvara identifierar genetiska varianter, förutspår deras klassificering baserat på integration av flera annotationskällor och genererar en variantrankning baserad på sjukdomsassociation. AION är en halvautomatiserad programvara för kliniskt beslutsstöd som hjälper vårdpersonal att fatta välgrundade medicinska beslut om kliniskt relevanta genetiska varianter och genetiska sjukdomar.
För närvarande avser AIONs indikation för användning testning av mendelska genetiska sjukdomar. Följaktligen är tolkning av varianter som kanske inte följer traditionella mendelska mönster för närvarande uteslutna från användningsindikationerna. Således är:
Se avsnittet ”Begränsningar av användning och kontraindikationer” för ytterligare information om dessa begränsningar samt om de åtgärder som vidtagits för att minska potentiella risker vid rapportering av denna typ av varianter.
Användning av AION utanför dess nuvarande indikationer för användning, kan leda till oväntat programvarubeteende och analysfel; det skulle göras under användarens eget ansvar.
AIONs indata är VCF-filer och det är användarens ansvar att tillhandahålla indata av hög kvalitet för att möjliggöra optimala utdataresultat för enheten. Se avsnittet om rekommendationer för VCF-format.
AION stöder NGS short reads-teknik på både Illumina och ION torrent, med de rekommendationer som anges per varianttyp, genomiska regioner och sekvenseringstekniker. Vi rekommenderar användarna att kontakta vårt supportteam om de använder andra typer av NGS-plattformar.
⚠️ Alla allvarliga incidenter som har inträffat i samband med enheten ska rapporteras till Nostos Genomics GmbH (se kontaktuppgifter nedan) och till den behöriga myndigheten i den EU-medlemsstat där användaren och/eller patienten är etablerad.
🗒️ För ytterligare information om säkerhet och risker med
enheten, vänligen kontakta oss Vår webbplats: Nostos Genomics – AI-driven
genetic analysis platform
Vår e-postadress: regulatory@nostos-genomics.com
AION es un software médico de diagnóstico in vitro (IVD) destinado a procesar cualitativamente datos de secuenciación de nueva generación (NGS) procedentes de pruebas genéticas. Este software de IVD basado en IA identifica variantes genéticas, predice su clasificación basándose en la integración de múltiples fuentes de anotación y genera una clasificación de variantes basada en la asociación con enfermedades. AION es un software semi-automatizado de apoyo a la toma de decisiones clínicas para ayudar a los profesionales sanitarios a tomar decisiones médicas informadas sobre variantes genéticas clínicamente relevantes y enfermedades genéticas.
Actualmente, la indicación de uso de AION se refiere a las pruebas de enfermedades genéticas mendelianas. En consecuencia, la interpretación de variantes que pueden no seguir los patrones mendelianos tradicionales está actualmente excluida de las indicaciones de uso. Por lo tanto
Consulte la sección de «Limitaciones de uso y contraindicaciones» para obtener más información sobre estas limitaciones, así como en las medidas adoptadas para mitigar los riesgos potenciales de la notificación de este tipo de variantes.
El uso de AION fuera de sus indicaciones de uso actuales puede causar comportamientos inesperados en el software, fallos en el análisis y será realizada bajo la responsabilidad exclusiva del usuario.
Los datos de entrada (input) de AION son los archivos VCF e información clínica del paciente (códigos HPO). Es responsabilidad del usuario proveer de datos de entrada (VCFs) de calidad para permitir la obtención de resultados óptimos del software. Para información adicional sobre el formato de los archivos VCF puede consultar la sección VCF format.
AION soporta la tecnología NGS short reads tanto en Illumina como en ION torrent, con las recomendaciones indicadas según las recomendaciones indicadas en la sección Variant type, genomic regions and sequencing technologies. Recomendamos a los usuarios que contacten con nuestro equipo de soporte si utilizan otro tipo de plataformas NGS.
⚠️ Cualquier incidente grave que se haya producido en relación con el dispositivo deberá notificarse a Nostos Genomics GmbH (véanse los datos de contacto más abajo) y a la autoridad competente del Estado miembro de la UE en el que esté establecido el usuario y/o el paciente.
🗒️ Para más información sobre la seguridad y los riesgos del dispositivo, póngase en contacto con nosotros a través de:
Nuestra página web: Nostos Genomics – AI-driven
genetic analysis platform
Nuestro correo electrónico: regulatory@nostos-genomics.com
AION è un software per dispositivi medico-diagnostici in vitro destinato all’elaborazione qualitativa dei dati di sequenziamento di nuova generazione (NGS) provenienti da test genetici. Questo software IVD guidato dall’intelligenza artificiale identifica le varianti genetiche, ne prevede la classificazione in base all’integrazione di più fonti di annotazione e genera una classificazione delle varianti basata sull’associazione con la malattia. AION è un software di supporto alle decisioni cliniche semi-automatizzato che assiste gli operatori sanitari nel prendere decisioni mediche informate in merito a varianti genetiche e malattie genetiche clinicamente rilevanti.
Attualmente l’indicazione d’uso di AION riguarda i test delle malattie genetiche mendeliane. Di conseguenza, l’interpretazione di varianti che potrebbero non seguire i tradizionali schemi mendeliani è attualmente esclusa dalle indicazioni d’uso. Pertanto:
Per ulteriori informazioni su queste limitazioni e sulle azioni intraprese per mitigare i potenziali rischi legati alla segnalazione di questo tipo di varianti, consultare la sezione “Limitazioni d’uso e controindicazioni”.
L’utilizzo di AION al di fuori delle sue attuali indicazioni d’uso potrebbe portare a un comportamento imprevisto del software e a un fallimento dell’analisi; ciò avverrebbe sotto l’esclusiva responsabilità dell’utente.
I dati di input di AION sono file VCF; è responsabilità dell’utente fornire dati di input di alta qualità per consentire risultati di output ottimali del dispositivo. Vedere la sezione dedicata al formato VCF per le raccomandazioni.
AION supporta la tecnologia NGS short reads sia su Illumina che su ION torrent, con le raccomandazioni indicate in base al tipo di variante, alle regioni genomiche e alle tecnologie di sequenziamento. Si consiglia agli utenti di contattare il nostro team di supporto se si utilizzano altri tipi di piattaforme NGS.
⚠️ Qualsiasi incidente grave verificatosi in relazione al dispositivo deve essere segnalato a Nostos Genomics GmbH (vedere i dettagli di contatto di seguito) e all’autorità competente dello Stato membro dell’UE in cui è stabilito l’utente e/o il paziente.
🗒️ Per ulteriori informazioni sulla sicurezza e sui rischi del dispositivo, si prega di contattarci
Il nostro sito web: Nostos Genomics – AI-driven
genetic analysis platform
Il nostro indirizzo e-mail: regulatory@nostos-genomics.com
| Date | IFU version | AION version | Changed page | Description of changes |
| July 28, 2026 | 57.2 | 3.20.0.2 |
Introduction Release Notes |
|
| June 25, 2026 | 57.1 | 3.20.0.1 |
Introduction Release Notes |
|
| May 18, 2026 | 57.0 | 3.20.0.0 |
Introduction API Usage ACMG/AMP Variant Classification Relevant Variants — CNVs ClinVar Classification and Conflict Resolution Artifact and Quality Filter Tags Annotation Processing Notes Data Sources Release Notes |
|
| March 11, 2026 | 56.1 | 3.19.0.1 |
Introduction Data Sources Release Notes |
|
| March 11, 2026 | 56.0 | 3.19.0.0 |
Introduction Data Sources Release Notes |
|
| February 16, 2026 | 55.0 | 3.18.0.0 |
Introduction Data Sources AION Variant Cards Release Notes |
|
| January 29, 2026 | 54.6 | 3.17.0.5 |
Introduction Release Notes |
|
| January 15, 2026 | 54.5 | 3.17.0.5 |
Introduction Release Notes |
|
| December 17, 2025 | 54.4 | 3.17.0.4 |
Introduction Release Notes |
|
| December 3, 2025 | 54.3 | 3.17.0.3 |
Introduction Release Notes |
|
| November 27, 2025 | 54.2 | 3.17.0.2 |
Introduction Data Sources Release Notes |
|
| November 25, 2025 | 54.1 | 3.17.0.1 |
Introduction Release Notes |
|
| November 25, 2025 | 54 | 3.17.0.0 |
Introduction Data Sources Release Notes |
|
| November 6, 2025 | 53 | 3.16.3.0 |
Introduction Release Notes |
|
| October 23, 2025 | 52 | 3.16.2.0 |
Introduction Release Notes |
|
| September 9, 2025 | 51.3 | 3.16.1.3 |
Introduction Release Notes |
|
| September 4, 2025 | 51.2 | 3.16.1.2 |
Introduction Release Notes |
|
| August 27, 2025 | 51.1 | 3.16.1.1 |
Introduction Release Notes |
|
| August 18, 2025 | 51 | 3.16.1.0 |
Introduction Data Sources Manual filtering - CNVs AION Results Variant type, genomic regions and sequencing technologies |
|
| July 2, 2025 | 50.1 | 3.16.1.0 |
Introduction Release Notes |
|
| June 27, 2025 | 50 | 3.16.0.0 |
Introduction Release Notes Data Sources |
|
| June 4, 2025 | 49.2 | 3.15.1.7 |
Data Sources Introduction |
|
| May 7, 2025 | 49.1 | 3.15.1.7 |
Introduction Release Notes |
|
| April 28, 2025 | 49 | 3.15.1.6 |
Introduction Safety Information Creating a Case VCF Format Variant type, genomic regions and sequencing technologies Data Sources Release Notes |
|
| January 15, 2025 | 48 | 3.15.1.0 |
AION Results Gene Coverage |
|
| December 19, 2024 | 47 | 3.15.0.0 |
Data Sources Overview of Cases Gene Coverage |
|
| November 26, 2024 | 46 | 3.14.0.0 |
Case Information AION Results AION Variant Cards |
|
| August 7, 2024 | 45 | 3.13.0.0 |
Data sources Overview of cases |
|
| July 25, 2024 | 44 | 3.12.3.0 |
Data sources Additional Variant Information AION Ranking General |
|
| July 19, 2024 | 43 | 3.12.2.2 | VCF Contents - secondary analysis best practices |
|
| July 3, 2024 | 42 | 3.12.1.1 | AION Database - variant statistics |
|
| June 24, 2024 | 41 | 3.12.1.0 |
Minimum IT
Requirements AION Database - variant statistics |
|
| May 30, 2024 | 40 | 3.12.0.0 | Data sources |
|
| May 16, 2024 | 39 | 3.11.1.0 |
Overview of cases AION Database - variant statistics AION Database - variant classifications Manual filtering - CNVs General |
|
| April 25, 2024 | 38 | 3.11.0.0 |
AION Ranking Manual filtering - CNVs Data sources |
|
| March 26, 2024 | 37 | 3.10.0.0 |
Introduction Generating a Report AION Ranking Additional Variant Information AION Database - variant statistics AION Database - variant classifications AION Database - onboarding |
|
| February 22, 2024 | 36 | 3.9.5.0 | Creating a Case |
|
| February 21, 2024 | 35 | 3.9.5.0 | VCF Format |
|
| February 8, 2024 | 34 | 3.9.4.0 |
Manual filtering -
Small Variants Manual filtering - CNVs |
|
| January 19, 2024 | 33 | 3.9.3.0 | General |
|
| January 16, 2024 | 32 | 3.9.3.0 | Advanced Filters |
|
| January 15, 2024 | 31 | 3.9.3.0 | AION ranking |
|
| December 20, 2023 | 30 | 3.9.2.0 |
Manual filtering -
CNVs Manual variant filtering - small variants Advanced filters |
|
| November 29, 2023 | 29 | 3.9.0.0 | Data sources |
|
| November 16, 2023 | 28 | 3.8.5.0 |
Generating a report |
|
| November 6, 2023 | 27 | 3.8.4.1 | Data sources |
|
| October 31, 2023 | 26 | 3.8.4.0 | Data sources |
|
| October 26, 2023 | 25 | 3.8.3.0 | VCF format |
|
| October 17, 2023 | 24 | 3.8.3.0 | Data sources |
|
| October 9, 2023 | 23 | 3.8.2.0 | Data sources |
|
| October 2, 2023 | 22 | 3.8.1.0 |
Case information Overview of cases Creating a case Analysing a case Run analysis Create a new case form |
|
| September 19, 2023 | 21 | 3.8.0.0 |
Data Sources VCF Format Creating a Case Creating a Case form Run analysis (Archived) Case Information AION ranking Internal variant database |
|
| September 1, 2023 | 20 | 3.7.1.0 |
VCF format Troubleshooting Manual filtering - CNVs |
|
| August 16, 2023 | 19 | 3.7.0.0 | Example use cases |
|
| August 11, 2023 | 18 | 3.7.0.0 |
VCF format Manual variant filtering - small variants Manual filtering - CNVs Data sources |
|
| July 14, 2023 | 17 | 3.6.0.2 |
AION - Instructions for
Use Safety Information |
|
| June 30, 2023 | 16.1 | 3.6.0.0 | VCF format |
|
| June 22, 2023 | 16 | 3.6.0.0 | Data sources |
|
| June 8, 2023 | 15 | 3.5.0.3 | VCF format |
|
| May 26, 2023 | 14 | 3.5.0.0 |
Create a new case form All pages IFU version changelog AION - Instructions for Use |
|
| May 24, 2023 | 13.1 | 3.3.1.0 | Overview of cases |
|
| May 23, 2023 | 13 | 3.4.0.0 |
VCF format Creating a case Case Information Analysis Information Data Sources Run analysis |
|
| May 23, 2023 | 12 | 3.3.1.0 | Variant type, genomic regions and sequencing technologies |
|
| May 18, 2023 | 11.5 | 3.3.1.0 |
AION - Instructions for
Use AION release notes |
|
| May 17, 2023 | 11.4 | 3.3.1.0 | AION - Instructions for Use |
|
| May 10, 2023 | 11.3 | 3.3.0.0 | AION - Instructions for Use |
|
| May 4, 2023 | 11.2 | 3.3.0.0 | Data sources |
|
| May 3, 2023 | 11.1 | 3.3.0.0 |
AION - Instructions for
Use Internal variant database |
|
| April 27, 2023 | 11 | 3.3.0.0 |
AION - Instructions for
Use AION release notes Annotations Data sources |
|
| April 26, 2023 | 10.2 | 3.2.4.0 | AION - Instructions for Use |
|
| April 21, 2023 | 10.1 | 3.2.4.0 | VCF format |
|
| April 14, 2023 | 10 | 3.2.4.0 |
AION - Instructions for
Use AION ranking Manual variant filtering - small variants Internal variant database Internal variant database details |
|
| April 12, 2023 | 9.1 | 3.2.4.0 | AION release notes |
|
| March 31, 2023 | 9 | 3.2.3.0 |
AION - Instructions for
Use VCF format AION requirements Variant type, genomic regions and sequencing technologies |
|
| March 27, 2023 | 8 | 3.2.3.0 | AION - Instructions for Use |
|
| March 16, 2023 | 7 | 3.2.3.0 |
Create a new case form AION release notes |
|
| March 14, 2023 | 6.3 | 3.2.2.0 | AION release notes |
|
| March 9, 2023 | 6.2. | 3.2.2.0 |
AION ranking AION release notes Overview of cases |
|
| March 2, 2023 | 6.1 | 3.2.1.1 | ACMG criteria |
|
| February 20, 2023 | 6.1 | 3.2.1.1 | AION release notes |
|
| February 6, 2023 | 6 | 3.2.1.0 |
VCF format AION release notes |
|
| February 3, 2023 | 5.3 | 3.2.0.4 | Create a new case form |
|
| February 1, 2023 | 5.2 | 3.2.0.4 |
AION release notes Example use cases |
|
| January 27, 2023 | 5.1 | 3.2.0.3 | Annotations |
|
| January 25, 2023 | 5 | 3.2.0.3 |
AION release notes Manual variant filtering - small variants |
|
| January 24, 2023 | 4.1 | 3.2.0.2 | AION release notes |
|
| January 23, 2023 | 4 | 3.2.0.2 |
AION - Instructions for
Use Create a new case form AION ranking VCF format Case results Anaysis information (archived) AION release notes ACMG criteria Technical requirements Example use cases Contact and legal information User account Creating a case |
|
| January 20, 2023 | 3.3 | 3.2.0.1 | AION release notes |
|
| January 12, 2023 | 3.2 | 3.1.0 | Case information |
|
| January 11, 2023 | 3.1 | 3.1.0 |
Example use cases Creating a case |
|
| December 23, 2022 | 3 | 3.1.0 |
AION - Instructions for
Use ACMG criteria Run analysis (archived) Additional variant information |
|
| December 22, 2022 | 2.3 | 3.1.0 |
AION - Instructions for
Use AION ranking AION release notes Manager screen |
|
| December 13, 2022 | 2.2 | 3.1.0 |
Create a new case form Annotation |
|
| November 23, 2022 | 2.1 | 3.1.0 |
Quality Overview Cases |
|
| November 15, 2022 | 2 | 3.1.0 |
AION - Instructions for
Use Case Information VCF Format Analysing a case Case results Literature & Sources Secondary analysis - best practices User account Creating a case |
|
| November 8, 2022 | 1.1 | 3.1.0 | AION - Instructions for Use |
|
| September 2, 2022 | 1 | 3.0.1 |
AION - Instructions for
Use Contact and legal information |
|
| June 2021 | 0 | 3.0.1 3.0.0 | NA |
|
Although VCF (Variant Calling Format) is a well-recognised standard file type in bioinformatics, variant calling pipelines might generate different kinds of VCF files. We strongly recommend performing variant calling following best practices, including processes such as variant normalisation and joint variant calling. Follow this link for further information.
Uncompressed and compressed VCF files are supported in AION. The file extensions must be .vcf or .vcf.gz.
To verify that your VCF files adhere to this standard, you can simply open them. VCF files can be opened by plain text editors (Notepad, TextEdit…). Even if they are initially recognised as Virtual Contact Files in some platforms, the files can be opened by text editors. In the case of compressed files, where the extension is .vcf.gz instead of .vcf, the files should be decompressed before opening.
All sequenced chromosomes from a sample need to be in the same VCF file, i.e. splitting VCF files into chromosomes is not supported.
ℹ️ Combined VCF files (including both small variants and
CNV/SV) are supported for the proband. Parents VCF files for
small variants need to be submitted as single-sampled VCFs
before uploading.
ℹ️ Genomic VCF files (gVCF) are not supported
Refer to Troubleshooting to see a list of general recommendations and error codes.
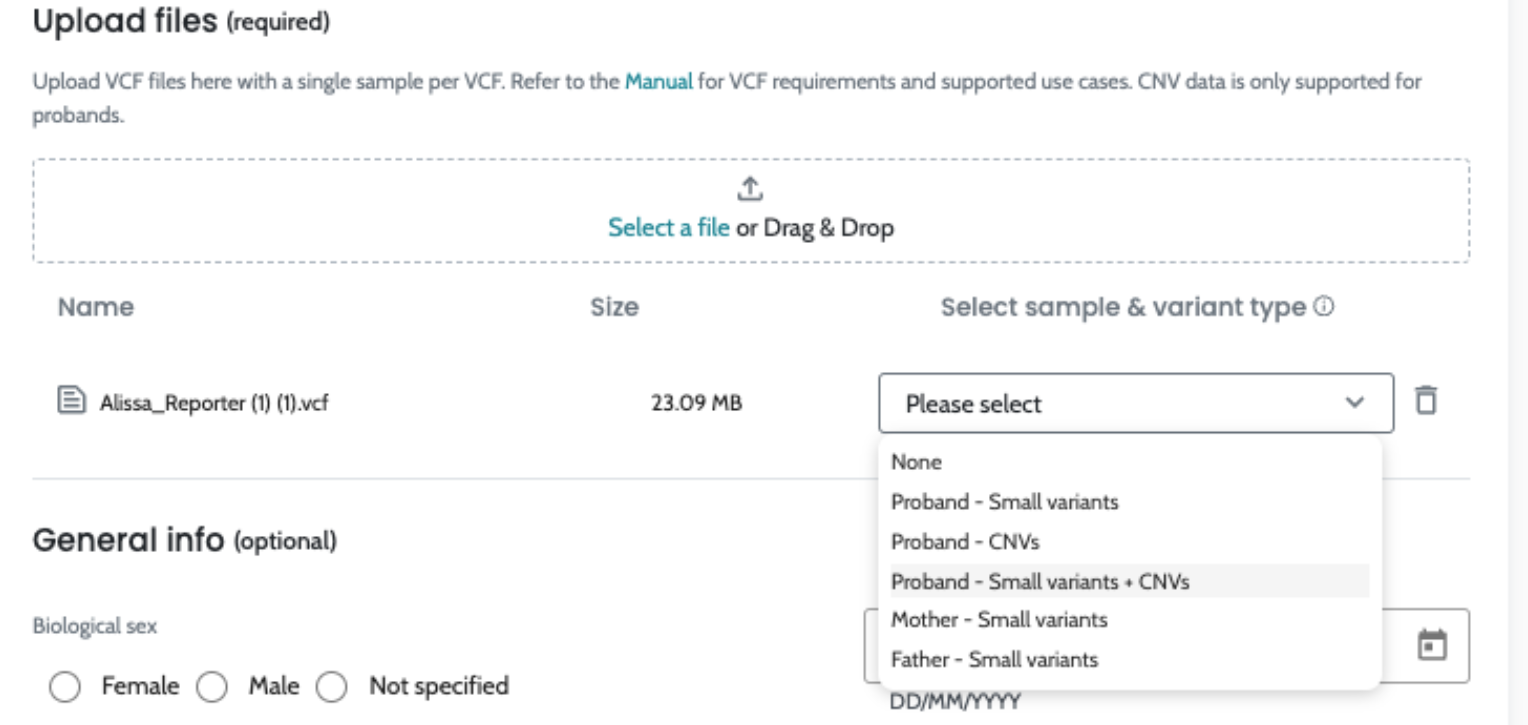
The information provided in this section is intended to assist users in preparing input VCF files for the AION platform; please note the following considerations regarding supported sequencing technologies and input VCF file formats:
The following is the minimum VCF file required by AION for small variants:
##fileformat=VCFv4.1
##FORMAT=<ID=AD,Number=R,Type=Integer,Description=“Allelic depths for the ref and alt alleles in the order listed”>
##FORMAT=<ID=DP,Number=1,Type=Integer,Description=“Approximate read depth (reads with MQ=255 or with bad mates are filtered)“>
##FORMAT=<ID=GT,Number=1,Type=String,Description=“Genotype”>
##contig=<ID=X,length=ZZZ,assembly=YYYY>
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT SampleID
chr3 239313 rs35603824 C CG 433899 PASS . GT:AD:DP 0/1:25,12:37The following is a checklist to make sure your VCF file is compatible with AION:
##fileformat=VCFv4.1##contig=<ID=X,length=ZZZ,assembly=YYYY>.CHROM, POS, REF, ALT, QUAL, FILTER, INFO, FORMAT, Sample ID
Before these column names, only one “#” symbol should be present as shown above
All columns except the Sample ID, which would change from one case to the next, should be written exactly as shown here
Sample ID column name should contain numbers, upper and lowercase letters. Symbols such as - and _ are supported, but others should be avoided along with special characters
For DP: AFDP
For AD: CLCAD2, VR, AO
If these data are not detailed properly in the header, the file might cause an error.
Format of FILTER, INFO, FORMAT specifications in the header:
##FILTER=<ID=ID,Description="description">
##INFO=<ID=ID,Number=number,Type=type,Description="description",Source="source",Version="version">
##FORMAT=<ID=ID,Number=number,Type=type,Description="description">AION supports CNV and SV data in VCF file format. Only CNVs
or SVs of type DUP or DEL are
supported. Only variants with VCF FILTER column value
PASS (or cnvLength in case of Canvas
& Dragen CNV) are annotated.
There are many supported pipelines and variant callers. The following is a shortlist of supported pipelines. Your pipeline may be supported even if it is not in this list or if it a combination of these callers:
For VCFs generated by Illumina tools, we additionally apply length based criteria and consider as relevant:
For a full description of the Relevant Variants tab, including sorting logic and how to switch to the All Variants view, see Relevant Variants — CNVs.
High-throughput sequencing technologies have enabled a large expansion of clinical genetic testing in the last decade and accurate variant calling in high-throughput data is a critical step before any interpretation process. There is a large variety of bioinformatic software and approaches for detecting genetic variants and different strategies to analyse clinical samples.
The quality of the input in AION influences the quality of the AION results. In this guideline we discuss the best practices for variant calling in clinical sequencing studies, with a particular emphasis on trio sequencing for inherited disorders. We do not want to conclude on which tools are the best ones, but to determine which strategies are the most recommended regardless of the bioinformatic tool selected.
As AION takes as input variant called files (VCF) generated by different variant callers (e.g. different customers or users), we have assumed VCF files are generated by following these best practices. It is also assumed that the format is supported by AION (see more details here)
There are dozens of bioinformatic tools to detect SNVs and INDELs, and countless more have been developed by researchers for internal use. Some of them are specialised in Whole Exome Sequencing (WES), other in Whole Genome Sequencing (WGS) or genomes, and many others in customised gene panels. Each variant caller is recommended for specific uses. Many benchmarking workflows have been performed and published so far (Krishnan et al 2021, Zhao et al 2020) comparing different variant callers on golden WGS trios available at the Genome In A Bottle (GIAB) consortium.
All variant callers can be applied to individual samples
after alignment and preprocessing are complete. It should be
noted that VCF files typically only contain entries for
positions that are different in a particular sample. This is,
when a variant is only detected in some samples but not others,
it is not clear whether the other samples are wild type for that
position (GT == 0/0) or simply did not achieve
sufficient coverage or other quality control for the variant
caller to make a call.
Joint variant calling - considering all samples simultaneously during variant calling offers several key advantages:
Copy Number Variation (CNV) variant calling using short-read sequencing technology, particularly in targeted panels or Whole Exome Sequencing (WES), presents several challenges.
The primary difficulty arises from the limited read length, which can complicate the accurate detection of CNVs, especially those spanning repetitive regions or having complex breakpoints. Short reads often fail to map uniquely to the reference genome in these areas, leading to ambiguous alignments and potential misidentification of CNV boundaries. Additionally, the depth of coverage is variable, further complicating the differentiation between true CNVs and sequencing noise.
To enhance the quality of VCF files and reduce artifacts and false positives, several strategies can be employed:
Ultimately, the quality of the CNV variants depends on the secondary analysis step. AION helps in interpreting CNVs, but its efficacy depends on the quality of the secondary analysis.
When implementing our pipeline to annotate, classify, and prioritize variants from any VCF file, AION has been developed assuming that VCF files followed bioinformatics best practices, allowing better sensitivity towards variant detection.
[1] Koboldt 2020 (Genome Medicine)
[3] Krishnan et al (BMC Bioinformatics) 2021
These instructions are not recommendations and should be strictly adhered to in order to ensure user safety.
Concerning VCF format and data requirements, please check the page dedicated to it.
AION is a cloud provided service with an evergreen client. The deployment and validation of all software and security updates are handled by Nostos Genomics and do not require any actions taken by the user or their IT team
AION is frequently updated to add functionality and increase security. Such updates may impact data integrity and analysis consistency. When such an update is scheduled, users are informed via the snackbar system with specific instructions on which actions to avoid at which time.
AION assumes best practices in secondary analysis. Specifically, we assume that trios and other pedigrees are jointly called.
AION supports automatic prioritisation of WES and WGS data containing:
⚠️ The current version of the AION user interface does not group and display CH pairs between a CNV and an SNP, due to a design limitation. However, according to recent publications (10.1056/NEJMoa2035790, 10.1038/s41431-022-01185-9, 10.1038/s41431-023-01312-0), it was found that only 2% of all diagnosed cases involved a CNV. Out of this, only a small fraction involved a CH pair between a CNV and an SNP. This means that not allowing to select CNV/SNP CH pairs will limit the ability to solve cases on less than 2%.
The supported regions for prioritisation are:
This means that with WGS data, no deep intronic variants will be automatically prioritised. Instead, the results will cover approximately the same regions for WES and WGS data.
AION supports gene panel data prioritisation as long as there is data within the supported regions.
AION is optimized for Next Generation Sequencing (NGS) data from short-read platforms, particularly Illumina technology. It is specifically designed to provide high-quality results when used with these technologies. However, the following limitations apply regarding other sequencing platforms:
For further details, including our control artifact list , and platform-specific artifact handling, please consult the relevant sections in the Instructions for Use (IFU, AION’s Limitations of use/ Contraindications).
AION’s compatibility with certain sequencing platforms may evolve as we gather more data and feedback from users. We encourage users to remain in close communication to ensure optimal performance with their specific sequencing setup.
All cases page lists all the cases that have been created. Each case can be identified by its Case ID number. It also shows the patient’s Sex, Age, Symptoms (HPOs used for the variant analysis), the analysis Start time, the User to which the case has been assigned, and the current Status of the case. A family icon before the Case ID indicates it is a trio case.
Hovering over Case ID, Sex, Age and HPOs fields will show the same values. This is important for smaller screens in which the values cannot be displayed fully due to screen size.
A User can be assigned to a Case by clicking on the Assign Case button, and then selecting one of the users associated to the account. A case can be unassigned by clicking on the “x” icon hovering over the user.
When you see View button in the case card, results are ready for review. Ability to delete a case can be found in the “…” selection
Case search functionality allows users to swiftly find the
case of interest when searching by CaseID/Case name. The search
feature allows user to select a single result, or filter the
whole case list with case ID exact or partial matches.
In future updates of the platform the search functionality will
be expanded to include search by case assignee and date of case
launch.
ℹ️ Find further visual support in the following clickable flow: Case Overview
The case status is shown in the top-right corner of the case card. It has several possible values:
Finished processing - a View status indicates that the automated variant analysis and ranking has been completed. By clicking on the tag for a case with ready results, you can access a dashboard that describes the variant analysis results for the specific case ID. This page consist of Case specific information, AION ranked list of variants as well as manual filtering view.
Processing - a Processing status indicates the automated variant analysis and ranking is being processed. The bar on the top of the case card visualises the progress.
Rejected - a Rejected status indicates that something is missing with the VCF file. Hover over with to get more details of the origin of the problem. If you still need clarification or further support check with the Nostos Genomics team.
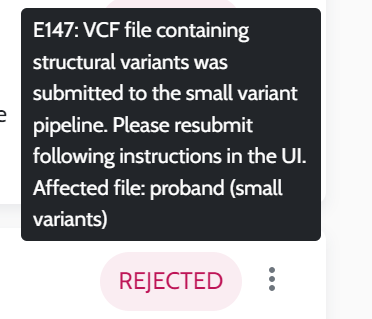
Updating - an Updating status indicates that reanalysis is running in case you have edited the patient symptoms from the case page. You may open the case page, but will not see any results until the reanalysis has finished.
Error - an Error status indicates that there are technical issues while processing the case. Hover to see more details. If you still need clarification or further support check with the Nostos Genomics team.
To facilitate finding or removing cases in a specific status on the general view, there is a checkbox filter that allows the user to apply a filter by status. The filtering allows multiple selection from the current cases status, including: processing, updating, error, rejected or finished processing.
The user can always see when the filter is active and the number of cases matching their search/filter. The filter can be easily removed/reset to show all the cases submitted in the account:
In case you want to remove a case from the list of cases, select Delete from the drop down menu accessible at the “…” icon.
Creating new cases is simple. Just go to the All cases section and select Create a new case.
 **
**
Create a new case form has now opened, allowing you to enter all needed information for the analysis.
When entering the Create a new case form, you will be asked to fill in information about the patient and analysis as well as uploading the VCF files for the case. You will be able to upload VCF files containing small variants (SNVs) and copy-number-variants (CNVs) for the patient. You will also have the opportunity to upload additional VCFs containing SNVs for the parents in case of a trio. General info allows you to input in sex, Date of Birth (DOB), symptoms in the Human Phenotype Ontology (HPO) and if needed, in silico panels to restrict the analysis to certain genes only.
See below for in-depth explanations of each section.
The case ID will serve as a reference to identify the case in the Results section. All formats are accepted but we recommend to use pseudonymized information in a standard format such as “NOS-200801-01” or “P20-1000A”, as this allows you to filter the cases efficiently.

The reference genome used during the secondary analysis is required here. AION also determines the reference genome based on the content of the VCF files as part of the quality control process. In case there is conflict in the user input and AION’s deduced reference, the user is informed.
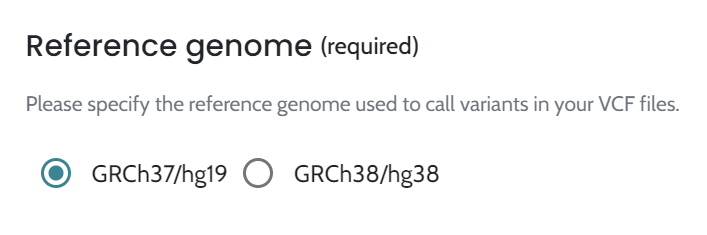
Upload files In this section, you can upload all the VCF files to be analysed as part of this case. The sample and variant type need to be selected through the dropdown. Please keep in mind that, in the current implementation, AION will only process familial cases if both Mother and Father’s VCF files are uploaded. Also, note that only small variants files are supported for the parents.
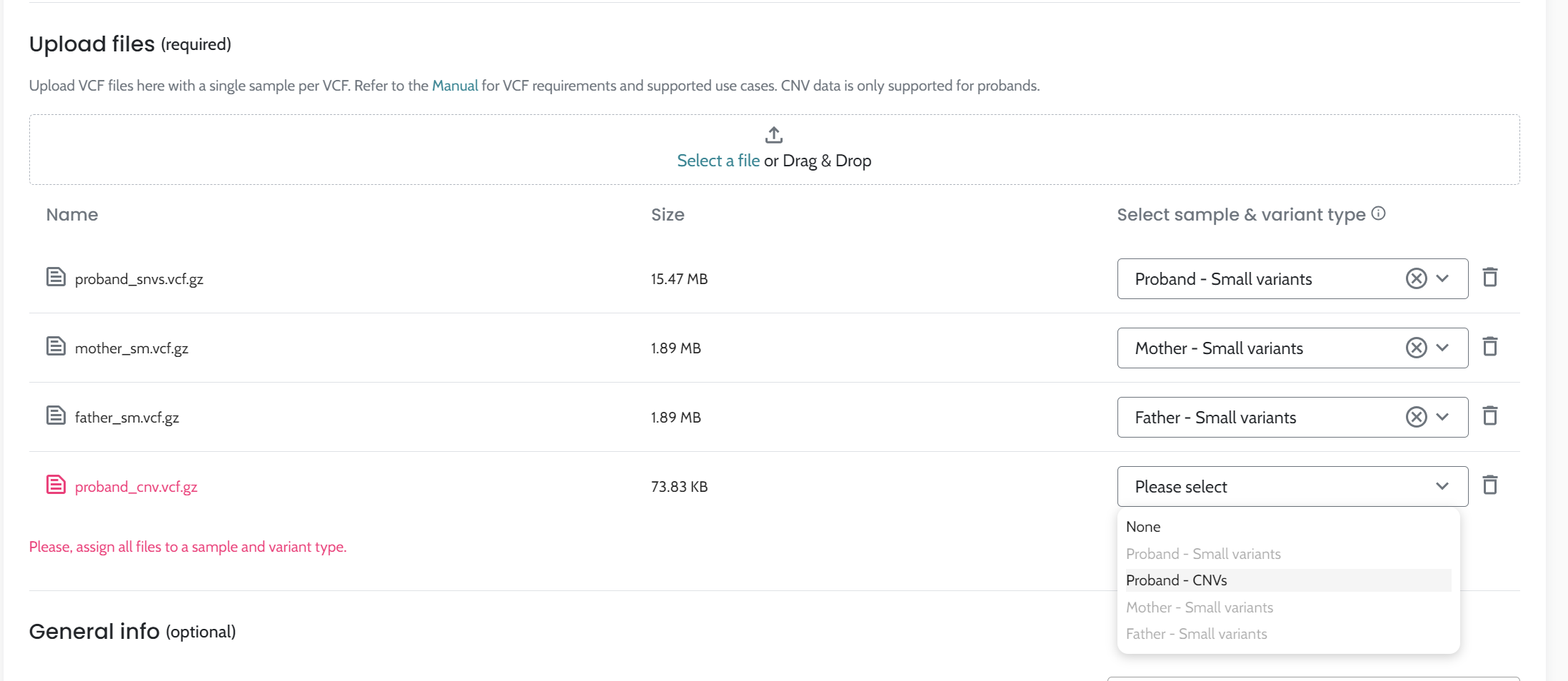
ℹ️ Notice that we support merged files containing both small variants and CNVs for probands. If you are submitting a merged file please select the option Proband - Small variants + CNVs in the drop-down to ensure both your small variants and CNVs are analysed.
You can optionally select one option (Female, Male, Not specified) and the DOB from the proband.
Currently, these values are exclusively used for your own case ID records and quality control.
Select as many symptoms/clinical features from the patient’s clinical history and presentation as possible. Symptoms can be inputted by
Searching for HPO terms and synonyms in the search bar
Pasting HPO IDs into the input area (e.g. HP:0000002, HP:0000003…)
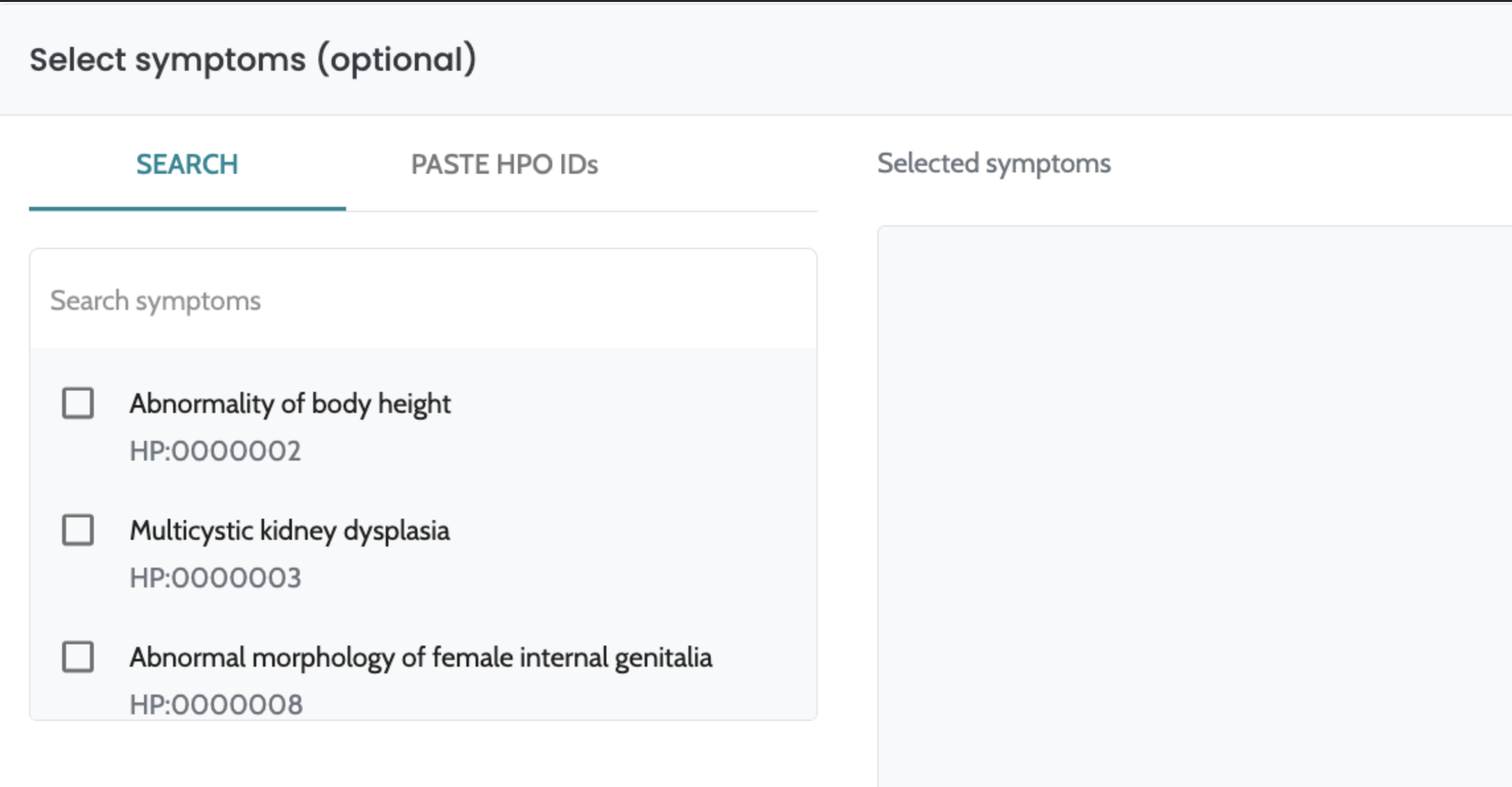
ℹ️ Including phenotypic information in HPO terms is optional but highly recommended. AION is designed to analyse variants considering symptoms and the clinical performance of the analysis may be affected. This is particularly relevant for CNVs, where providing HPO data greatly improves the accuracy of variant ranking, ensuring more reliable and meaningful results.
The selected HPO terms should reflect the patient’s phenotype as much as possible. It is important to note that the list of included HPO terms impacts AION automated filtering and ranking of variants relevant to the patient’s disease. The selected HPO terms can include clinical features, laboratory abnormalities (such as Abnormal circulating metabolite concentration - HP:0032180) and test results (Abnormal EKG - HP:0003115). The HPO browser is a useful resource when trying to search for the patient’s symptoms.
Once you have selected a symptom, AION will also provide a list of suggested related symptoms that might help refining the patient phenotype.
Users can choose to restrict the results to specific genes to focus on selected panels and/or to avoid secondary findings. Applying in silico panels effectively defines areas of interest within the supported regions.
To do so, AION enables filtering of specific genes in 3 different ways, which can be applied separately or in combination:
Applying gene panels: select one or more
gene panels, containing genes related a specific disease or
health condition. Each condition has been curated into 4
different lists (Candidate Genes, Priority genes, Priority and
Secondary genes, and Secondary genes) so that user can select
the strength of the evidence backing up the relationship between
genes and a specific condition.
There are special panels also to exclude from the analysis the
78 genes whose variants are recommended to report as secondary
findings by ACMG (Miller
et al., Genetics in Medicine 2022), or to only focus on
these 78 genes (search for “secondary findings” to apply
them).
Search genes: select individual genes.
Paste genes: paste a list of genes separated by commas.
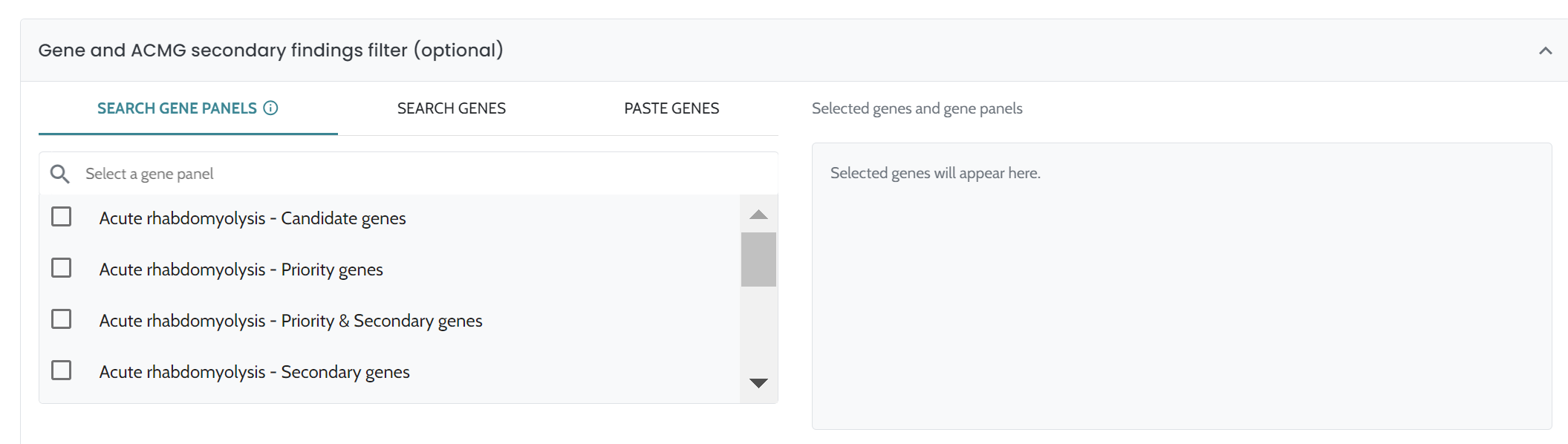
In case of a trio analysis, fill the affectedness status of the parents.
After filling in the case submission form, selecting “Run Analysis” will submit the case and start the process of data uploading, variant annotation, classification and ranking. You will be kept on upload screen while the data is being uploaded. After this, you are redirected to All cases and can continue working on other cases while the newly submitted case processes.
Once the analysis is done, a list of prioritized and classified variants for the submitted cases will show up in the All cases page. When a case shows a “View” button instead of processing status, you can open it to see the list of prioritized and classified variants.
ℹ️ Find further visual support in the following clickable flow: Creating a Case in AION
Case information is shown when opening a case page from the case listing and the case information page consist of:
Case header: always shown on case page when scrolling through the results, visualising basic case identifiers and information as well as reporting functionality
Information sections: divided into Clinical and Analysis information
Download results button: allows downloading the results in a tsv format.
Fixed case header contains the following information and actions:
Case ID
Sex and Age of the patient
Edit the case summary - Write your case report summary. This summary will be shown in the exported report. Case summary is needed in order to generate a report.
Generate a report - Once you mark one/several relevant variant(s) and add a report summary, you can download a PDF with a summary of the case here.
Date of submission & submitter - The date for the submission of the case to AION, and the user who submitted the case
Assign case - Allowing you to assign the case for yourself or a colleague
Version history - you can visualize the version history of the case.

⚠️ Editing the patient symptoms will launch a re-analysis with the new HPOs and results will change. Old results will no longer be available for review.
Case type (singleton/trio)
Parents affected status in case of a trio.

ℹ️ Find further visual support in the following clickable flow: Editing Symptoms / HPO terms
Uploaded data for this case. Original files names can be reviewed through “Show” button
The reference genome version information: The reference genome used by AION analysis pipeline for analysing the results. This should match the input reference genome of your files. The supported reference genomes are GRCh38/hg38 and GRCh37/hg19. Reference: Exome variant discrepancies due to reference-genome differences
The in silico gene panels used in the analysis
AION DB (database) statistics refreshing: shows date and time in which the statistics in AION DB have been been updated. The “Refresh” of the statistics can be manually performed every 30mins for cases with GRCh37/hg19 reference genome. The refresh action can is active if there are cases submission after the submission of the current case. For more information visit AION variant statistics.
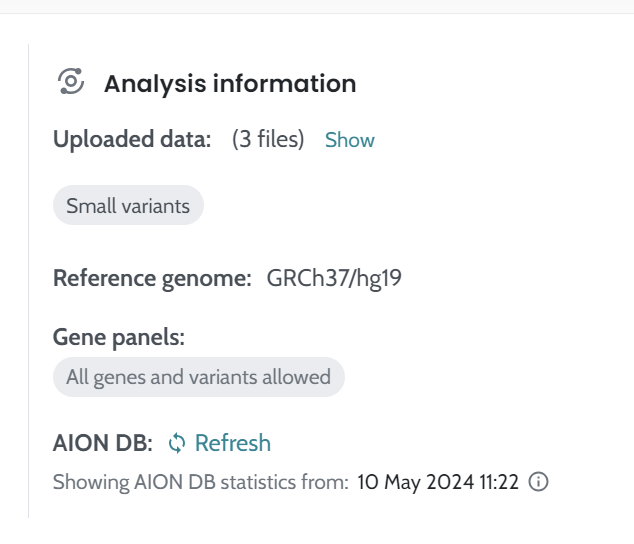
ℹ️ If the name of the vcf file(s) submitted for the case analysis is detected to be duplicated the AION DB would not be showing in the case Analysis information section.
Gene based coverage data extracted from the BAM file can now be uploaded to AION in tsv format. This data is then transformed by AION in a user friendly xlsx file allowing easy visualisation, sorting and filtering for instance by a list of genes of interest. The xlsx file is available for download for all cases where this data exist under the download results dropdown:
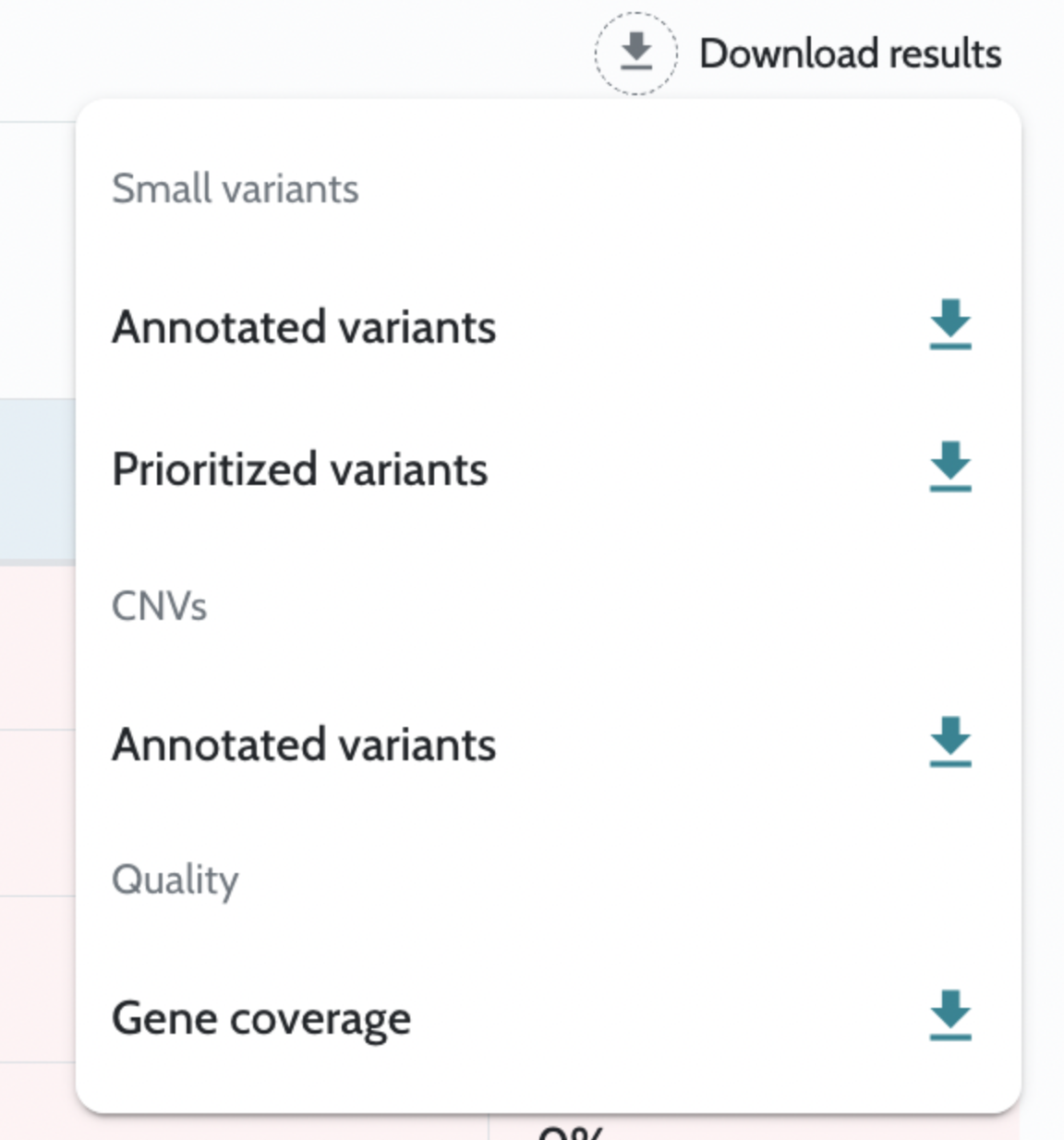
Note! The definition of region to consider for a gene to be fully covered, depends on the analytics pipeline producing the data. Thus, this might vary between samples and customers.
If you are using Nostos Genomics secondary analysis for the creation of the gene coverage data the region to define for a gene to be fully covered depend on the sample type and is the following;
Annotation file: table containing all the annotated variants submitted to AION in tsv format.
Prioritizer file: table containing the prioritized variants available in the AION results tab. This file (tsv) also contains symptoms (and their corresponding HPO codes) used to perform the prioritization.
Annotated variants - VCF file name: table containing the annotated CNV pipeline results. CNV results are sorted by relevance, showing the most relevant candidate CNVs at the top of the downloadable file.
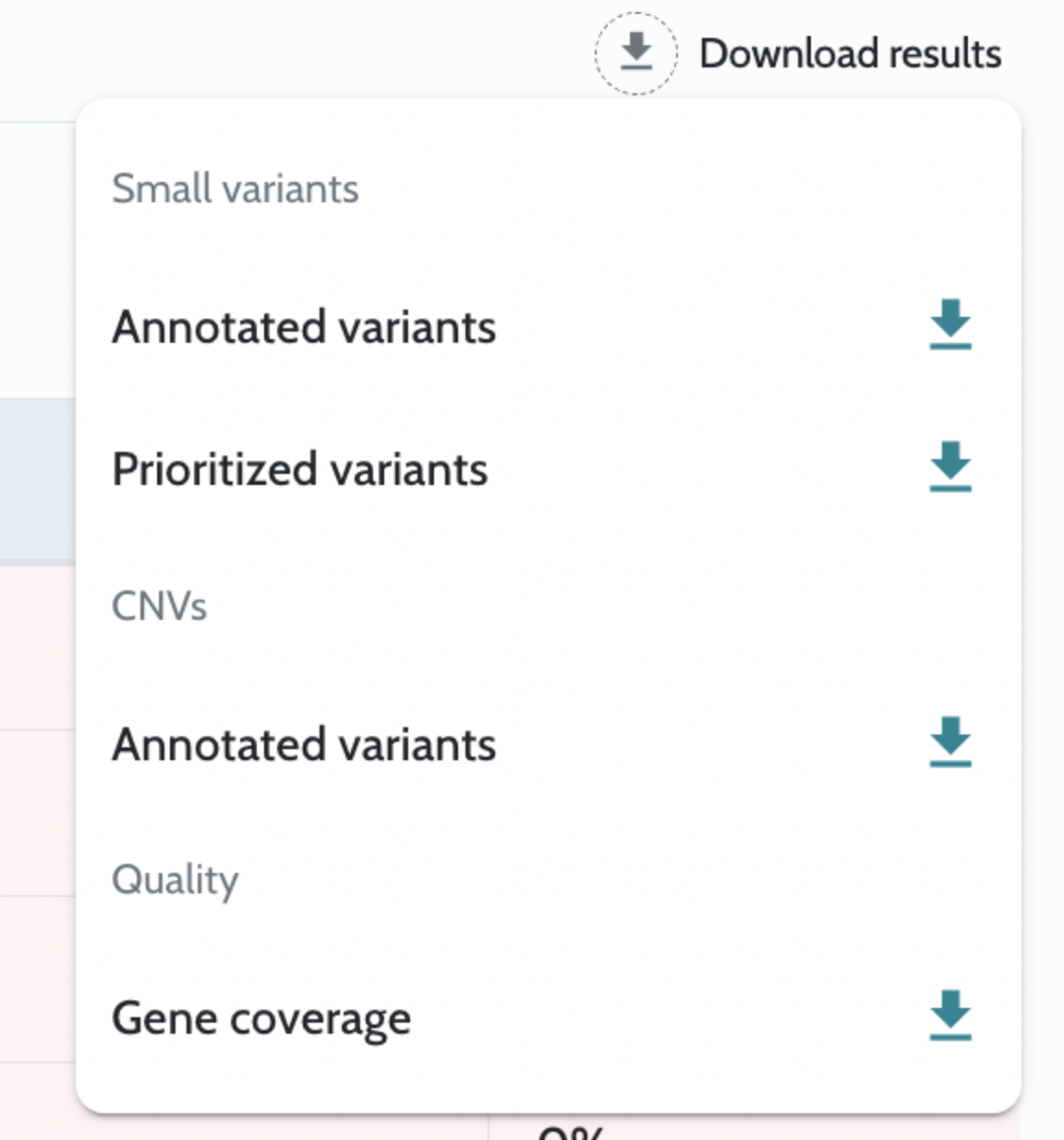
When reviewing CNV analysis results, AION distinguishes between All Variants and Relevant Variants. The Relevant Variants tab displays a curated subset of CNVs that AION has automatically prioritised as most likely to be clinically significant.
Relevant variants are a subset of all CNV variants selected by AION based on several inclusion and sorting criteria, designed to surface the most clinically actionable CNVs first. Not every variant in the VCF will appear in this tab; the goal is to reduce the manual review burden by filtering out likely benign or low-evidence calls.
A CNV variant is included in the Relevant Variants tab if it meets any of the following criteria:
For VCFs generated by Illumina tools (Canvas, DRAGEN CNV, Manta, DRAGEN SV), additional length-based criteria apply:
Relevant variants are sorted by a combination of ACMG classification, Phenotypic score, and ACMG score — ensuring that the most clinically significant and evidence-supported variants are shown at the top of the list.
ℹ️ The Relevant Variants tab is designed as a starting point for CNV review. If you need to review all CNVs without filtering — for example when investigating borderline variants or performing a comprehensive audit — switch to the All Variants tab, which displays every annotated CNV without exclusion.
All variants from AION results (Smoking guns and AION Clues) and Manual filtering - small variants tabs can be added to the PDF report. This happens by selecting the “Report” symbol in the variant card:
⚠️ CNV variants cannot be added to the report. This is a known limitation currently being worked on.

To generate and download a report you need to have at least one variant selected, as well as have a report summary written. Reports are available in English or Spanish.

⚠️ Currently there is no possibility for the user to select the final classification of the variant for the report, but we refer to automatised ClinVar and ACMG classifications for variant pathogenicity and the Previous classifications (institution database), which is the manual classification label given by the user. In certain situations, these might be in conflict and cause a risk of misinterpretation. This is a known issue currently being worked on.
If you need specific report functionality, contact support@nostos-genomics.com so your requirements can be supported.
ℹ️ Find further visual support in the following clickable flow: Generating a report in the AION Platform
Gene coverage information can be downloaded as an excel (xlsx) file from the user interface (UI) as mentioned in the relevant section in the Case Information page. In addition, the information is also available in a specific tab in the UI.
The view by default includes the columns with the information for depth at 5X, 10X, 20X, 30X, 50X 100X and 150X, the Min Depth, Max Depth and the Average depth for each gene, and default sorted by Gene column. Non-coding genes are labeled with an asterisk (*) to raise awareness to the user:
The user can reduce the number of columns visible by unselecting any depth information that is not relevant to be considered in the current analysis, filter by any values for a specific depth and change the sorting criteria. Any changes in the sorting or filtering applied will be showing on the top of the coverage information table as shown in the screenshot below:
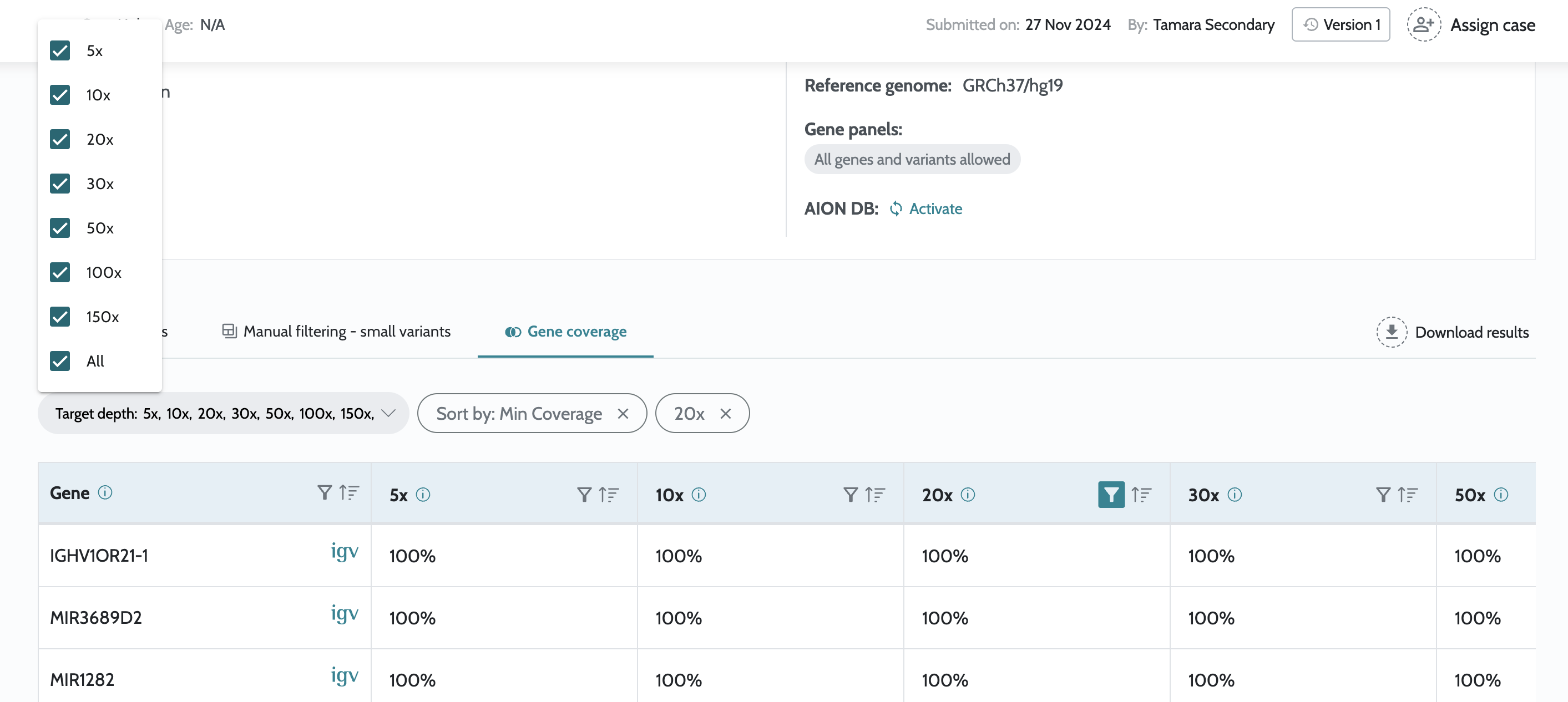
Regarding coverage values, it is important to consider that gene coverage is influenced by the definition according to the secondary pipeline used to produce the data. In the case of using Nostos Genomics secondary pipeline the gene definition for coverage calculation is as follows:
Internal annotations are used by the pipeline for prioritisation, variant classification, quality control, and preliminary filtering.
| Data source | AION version hg19 | AION version hg38 | Citations |
| BP1 gene list | Based on ClinVar 18/04/2026 | Based on ClinVar 18/04/2026 | Richards, Sue et al. 2015 |
| Coding region BED |
RefSeq canonical transcripts (2024-09-07) ±50 bp; includes ClinVar (18/04/2026) pathogenic regions ±3 bp |
RefSeq canonical transcripts (2025-08-06) ±50 bp; includes ClinVar (18/04/2026) pathogenic regions ±3 bp |
Relevant for MANE: Morales, Joannella et al. 2022 |
| Global frequent artifact blacklist | Internal computation - v2 | Internal computation - v1 | - |
| Hotspot definition | Based on ClinVar 18/04/2026 | Based on ClinVar 18/04/2026 | Richards, Sue et al. 2015 |
| NMD | Based on Refseq (2024-09-07) | Based on Refseq (2025-08-06) | Torene, Rebecca I et al. 2024 |
| PP2 gene list | Based on ClinVar 18/04/2026 | Based on ClinVar 18/04/2026 | Richards, Sue et al. 2015 |
| PVS1 gene list | Based on ClinVar 18/04/2026 | Based on ClinVar 18/04/2026 | Richards, Sue et al. 2015 |
| Repeat definition | RepeatMasker from UCSC 2020-02-20 | RepeatMasker from UCSC 2022-10-18 (hg38) | RepeatMasker Jurka, J et al. 2005 |
| Uniprot functional domains | 06/2020 | 05/2023 | Uniprot |
| Whitelist of variants to keep with high gnomad freq | Internal computation - v1 | Internal computation - v1 (lifted-over) | - |
| Source | hg19 | hg38 | Citation |
| AION classification | Internal ML model - v1 | Internal ML model - v1 (lifted-over) | - |
| BLOSUM62 | BLOSUM62 | BLOSUM62 | Henikoff S and Henikoff JG. 1992 |
| ClinVar | 18/04/2026 | 18/04/2026 | Landrum MJ et al. 2018 |
| dbscSNV | v1.1 | v1.1 | Jian X et al. 2014 |
| GnomAD | 2.1.1; 4.1.0 (liftover) | 3.1.2; 4.1.0 | Chen, Siwei et al. 2024 |
| Grantham scores | From paper | From paper | R. Grantham 1974 |
| Pangolin splice scores | - | Precomputed scores from Zenodo record 15649338 | - |
| Pfam domains | ucscGenePfam hg19, 2025-09-26 | ucscGenePfam hg38, 2025-09-26 | UCSC Genome Browser |
| PhastCons | UCSC phastCons46 | UCSC phastCons100 | Siepel A et al. 2005 |
| PhyloP | UCSC phyloP46 | UCSC phyloP100 | Pollard KS et al. 2009 |
| RefSeq transcripts | 27/10/2024 | 06/08/2025 | RefSeq: NCBI Reference Sequence Database |
| REVEL | v1.3 | v1.3 | Ioannidis et al. 2016 |
| VEP (Variant Effect, Transcript, Gene, dbscSNV, SIFT, PolyPhen) | 115.2 | 115.2 | McLaren W et al. 2016 |
| Source | Version | Citation |
| DDG2P | Downloaded 17/11/2025 | DDG2P |
| GenCC | Downloaded 17/11/2025 | GenCC |
| HGNC | Downloaded 17/11/2025 | HGNC |
| HPO | Downloaded 17/11/2025 | HPO |
| MedGen | Downloaded 17/11/2025 | MedGen |
| Mondo | Downloaded 17/11/2025 | Mondo |
This section describes how AION derives or processes certain annotation fields that are not sourced directly from an external database but instead computed internally from reference data.
PolyQ (polyglutamine) regions are defined at the protein level and used by AION to annotate variants that fall within or adjacent to repetitive glutamine tracts — a feature relevant to several neurodegenerative Mendelian diseases.
AION identifies PolyQ regions using the threshold described in The Protein Structure Context of PolyQ Regions (PMCID: PMC5268486): a region qualifies when at least 8 glutamines (Q) are present within a sliding window of 10 amino acids, allowing up to 2 non-Q residues within that window. Overlapping qualifying windows are merged into a single PolyQ region.
RNA-level confirmation is based on consecutive
CAG codons, and a transcript-level tract is
reported only when its translated glutamine run falls within one
of the protein-level PolyQ regions identified above.
The annotation output includes two related fields per variant:
polyQ_window_* — the exact
qualifying protein window that triggered the PolyQ call.polyQ_* — the broader merged
protein PolyQ region encompassing the qualifying window.Nonsense-mediated mRNA decay (NMD) is a surveillance pathway present in all eukaryotes. Its main function is to reduce errors in gene expression by eliminating mRNA transcripts that contain premature stop codons.
AION computes NMD escape boundaries from CDS-bearing exons using the following logic:
before-last-exon and last-exon
regions.single_exon region clipped to the CDS-covered part
of that exon, plus 2 bp on each side.NMD boundaries are computed from RefSeq transcript annotations. See the Shared annotation sources table for the RefSeq version used.
ClinVar aggregates variant classification submissions from many independent laboratories and clinical centres. In many cases, different submitters reach different conclusions about the same variant. Rather than displaying the raw CLNSIG field from ClinVar (which may reflect these conflicts as a combined string), AION re-derives a single consolidated classification from the underlying submission counts — referred to as ClinVar Nostos.
This approach is designed to be conservative with conflicts: where the evidence is genuinely ambiguous, AION will return VUS rather than resolving the conflict in either direction.
ℹ️ De novo variants — where the ClinVar origin field includes “de novo” — are always assigned Pathogenic regardless of any conflicting submission counts. This reflects the high clinical weight given to confirmed de novo occurrences in pathogenicity assessment.
⚠️ The ClinVar Nostos classification is a computational consolidation of available submission data and does not replace expert review of individual ClinVar submissions. For variants where the classification has significant clinical impact, we recommend reviewing the full submission history directly on the ClinVar website.
AION results contains the variants automatically identified by AION as the most relevant in the case. To address the diverse needs of clinical diagnostics and research exploration, AION provides two different lists of prioritised variants;
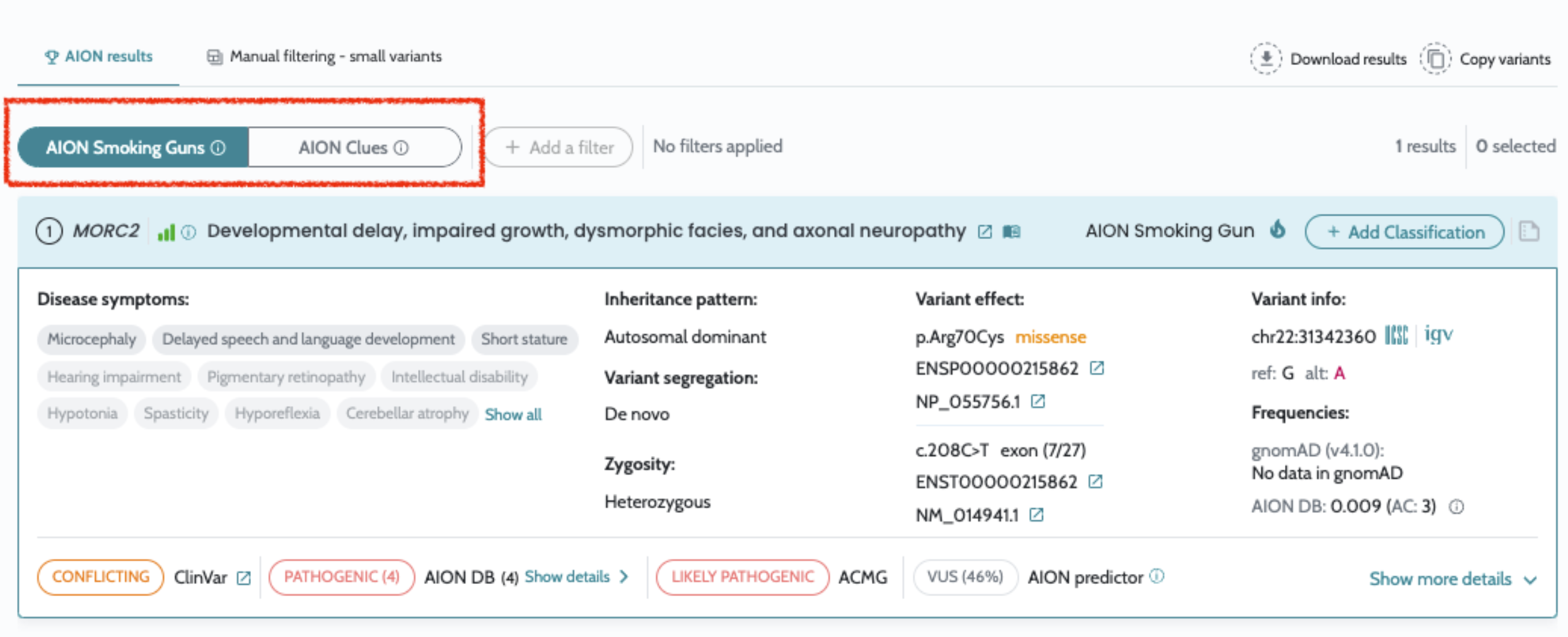
AION Smoking Guns is the best place to start analysis. It contains strong diagnostic candidates fulfilling all the below criteria:
ℹ️ Notice: All ClinVar 3 or 4 star P/LP variants are always picked up as AION Smoking guns.
AION Smoking Guns might not be present for your case. When this happens it means that AION has not identified a very strong and clear disease causing candidate for your case. This might be due to a missing patient phenotype description, and adding that might help.
In all your cases AION will still be providing you with AION Clues, to support you to start your analysis with potential candidates for your case.
⚠️ Notice: AION Smoking Guns and AION Clues are currently considering Small Variants only. For relevant CNV variants, you still need to analyse the Manual filtering - CNVs tab separately.
AION Clues are the next best place to continue the analysis, if you have already reviewed AION Smoking Guns. It contains potential diagnostic candidates that show evidence of pathogenicity, including the AION Smoking Guns and other candidates that are might be lacking some evidence or the evidence is not strong enough for claiming its position as a AION Smoking Gun.
Evidence of pathogenicity is evaluated based on molecular, phenotypic, segregation and database evidence in a less stringent way than with AION Smoking Guns.
AION Clues provides a wide range of options ideal for cases where the solution is not as clear and are available also when no patient phenotype is entered.
ℹ️ For further information on the variant information check the section on AION variant card and Additional variant information
The following annotations are shown for each analyzed and ranked variant:
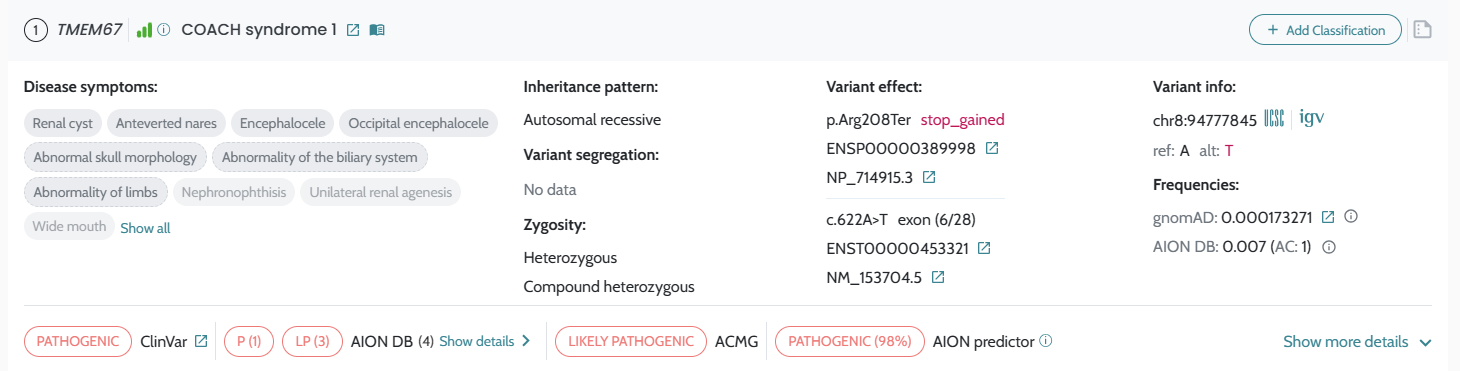
For each prioritized variant, either in AION Smoking Guns or AION Clues list, shows the following information is shown:
AION rank AION rank is the main output of our platform. Our prioritization of classified variants in AION is based on obtaining the best matches between 1) patient HPO terms, and 2) HPOs related to rare diseases, associated to genes in which candidate pathogenic variants have been found. Variants are considered pathogenic based on a combination of ClinVar, our implementation of ACMG/AMP guidelines, and ML-based classification values.
Variants are ranked according to a patient-disease phenotype similarity, associated modes of inheritance for each gene, as well as co-segregation information (when parents VCF files have also been uploaded) and other metrics such as variant tiering. The bigger the phenotype similarity, the more confident we are that a variant or variants (compound heterozygous for example) can explain the patient’s disease. The list of HPOs inputted for a Case is taken into account to calculate the p-value for a variant, and variants are ranked/prioritized according to the p-value. Therefore, it is important to provide as much clinical information as possible from a patient, in the form of HPOs, to obtain accurate phenotype similarity values for variant prioritization.
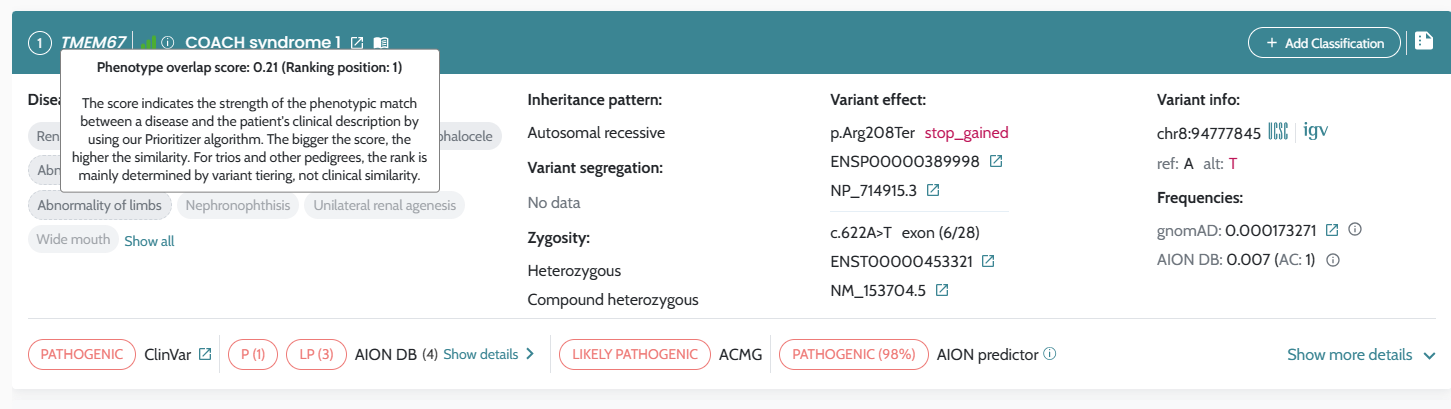
Gene where the variant is located and related disease are shown in the header of the card, with confidence of the gene-disease-moi association and link to the source.
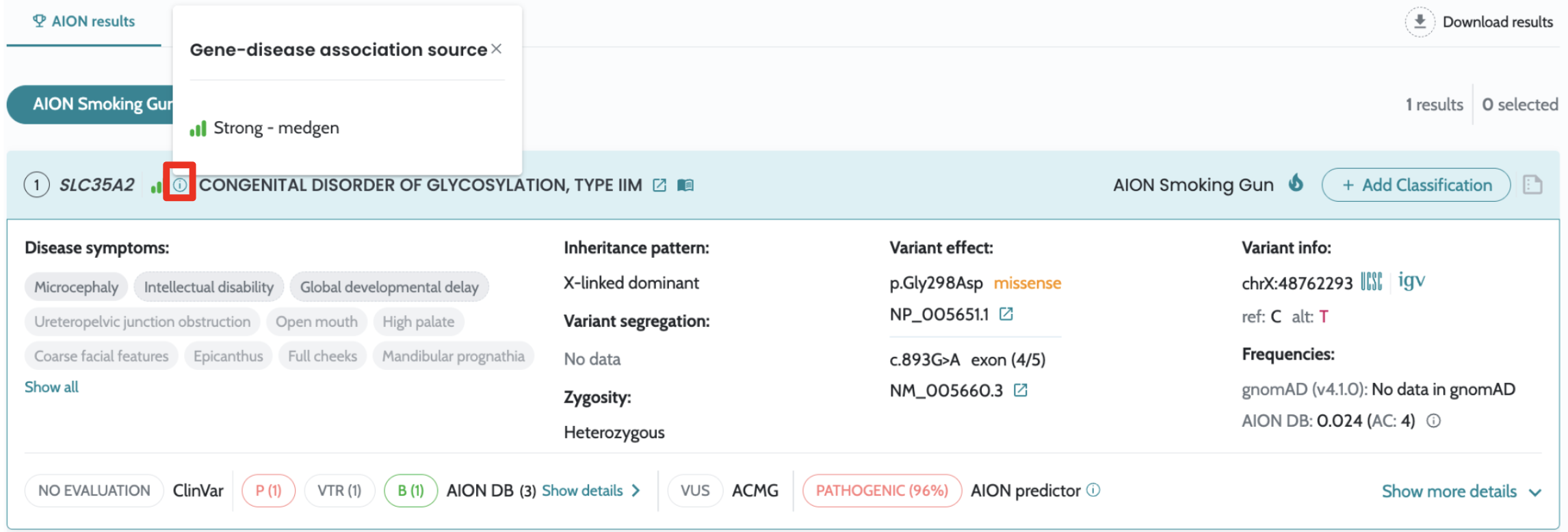
ℹ️ Notice that gene–disease associations are initially sourced from external databases. However, key attributes—including mode of inheritance, association strength (confidence level), disease name, and overall validity—are subsequently manually curated through a detailed review of the scientific literature. This rigorous curation process ensures high data quality, accuracy, and clinical reliability.
Classification Available classification for a variant is displayed according to 3 different sources:
ClinVar: ClinVar database variant classification. If a variant has been described in ClinVar, clicking on the link will open the ClinVar webpage for the described variant, with extensive information on variant evaluation, submitters, etc. A variant can be classified as Pathogenic (P), Likely Pathogenic (LP), Variant of Uncertain Significance (VUS), Likely Benign (LB) or Benign (B), among other values. Conflicting interpretations are also displayed (CI). When the variant has not been described in ClinVar “NO EVALUATION” is displayed.
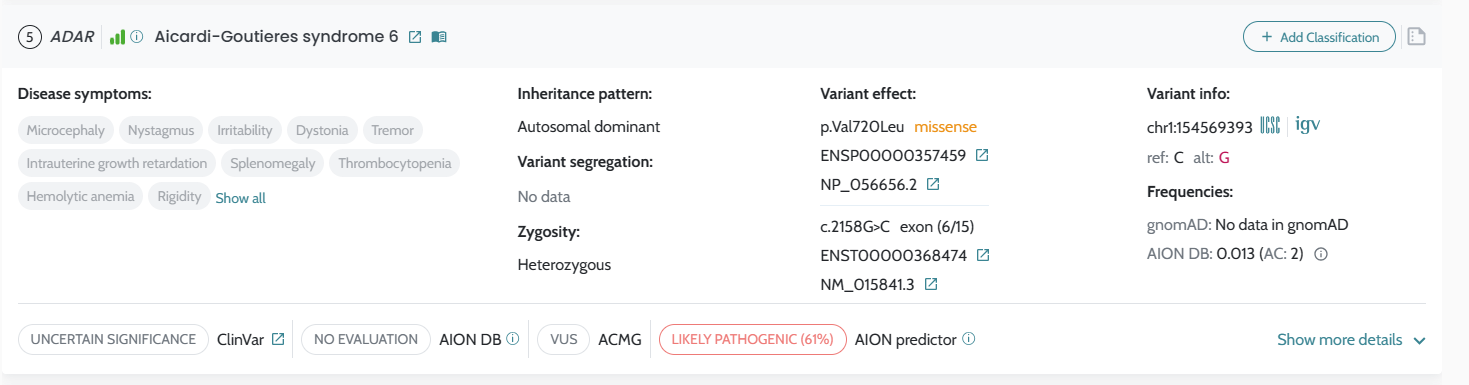
Notice that the updated category “Conflicting classifications of pathogenicity” will remain to be shown as “Conflicting interpretations of pathogenicity” in the user interface in order to facilitate the user filtering by this conflicting category in a uniform and consistent way under the same category.
AION DB: Previous classifications done by users from your institution on this variant will be shown.
ACMG: ACMG/AMP variant classification has been implemented in AION following the ACMG/AMP variant classification standards. The application of all criteria can be modified by the user, changing the overall ACMG/AMP classification. A variant can be classified as Pathogenic (P), Likely Pathogenic (LP), Variant of Uncertain Significance (VUS), Likely Benign (LB) or Benign (B). Conflicting interpretations are also displayed (CI). VUS variants are further sub-classified on a scale from “Ice Cold” (minimal evidence) to “Hot” (strong but inconclusive evidence) to provide additional clinical context. See ACMG/AMP Variant Classification for full details.
AION applies ACMG/AMP guidelines taking only into account the criteria that can be deduced from the VCF files. That means that some variants may be initially classified as VUSs if additional criteria that are not related directly to the variant are not assessed manually by the user.
AION Predictor: This score displays the output of our Classifier Machine Learning-trained algorithm. The Classifier uses multiple variant properties and its underlying genomic region to classify a variant, such as evolutionary prediction scores, splicing prediction scores or variant frequency in healthy population databases as an input. Based on this score, the algorithm provides a predicted variant classification:
0% - 15% = Benign
15% - 40% = Likely benign
40% - 60% = VUS (Variant of Unknown Significance)
60% - 85% = Likely pathogenic
85% - 100% = Pathogenic
Rescued = Variants brought to the prioritized variants list due to the fact that they are a partner with another variant already prioritized (Compound heterozygous).
Disease symptoms are shown, highlighting their overlap with the patient’s symptoms. Total phenotypical overlap is shown in dark grey, while partial overlap shows an additional dashed line around the symptom.
Inheritance pattern. This field displays the OMIM ID candidate genetic disease as well as the associated inheritance pattern/s, as prioritized by AION.
⚠️ For diseases with an X-linked (XL) mode of inheritance without specifying whether it is XLD or XLR, the inheritance mode is considered as XLD to be more conservative and prevent from missing potentially relevant variants in heterozygosis.
Segregation: Segregation in trio family. If no parents are uploaded, all variants will display “No data”. Also a possible AION detected compound heterozygosity with another variant will be highlighted here.
Zygosity: Patient’s variant zygosity (heterozygous, homozygous), as a reflection of genotype data in Annotations. Reference and alternative alleles.
Variant effect are shown along with:
cDNA change, exon/intron location, and reference transcript (Ensembl).
Protein change, variant type (e.g missense, splice region variant) and reference transcript (Ensembl). Variant type is colored in red, orange or blue to denote the variant type impact on protein (red: high, orange: moderate, blue: low). Clicking on Ensembl transcripts redirects users to Ensembl webpage, to show several informations related to each protein/mRNA isoform.
Location: Chromosome and Genomic reference location (in the reference genome indicated in analysis information section.
Frequency displays the alternate allele frequency for the variant of interest.
If the variant is found in the internal variant database, then information will be shown related to the internal frequency and past interpretations. See more details in the internal variant database page.
ℹ️ For further details on the variants information and annotation please visit Additional variant information
All variants have a link to the IGV desktop application to allow visual exploration of the genomic data. To be able to visualize the data you should have the IGV desktop application open with the corresponding reference genome and relevant BAM files. Additionally, port listening must be enabled and configured to the default port (60151) in the Advanced tab of the View > Preferences window (refer to IGV for more information).
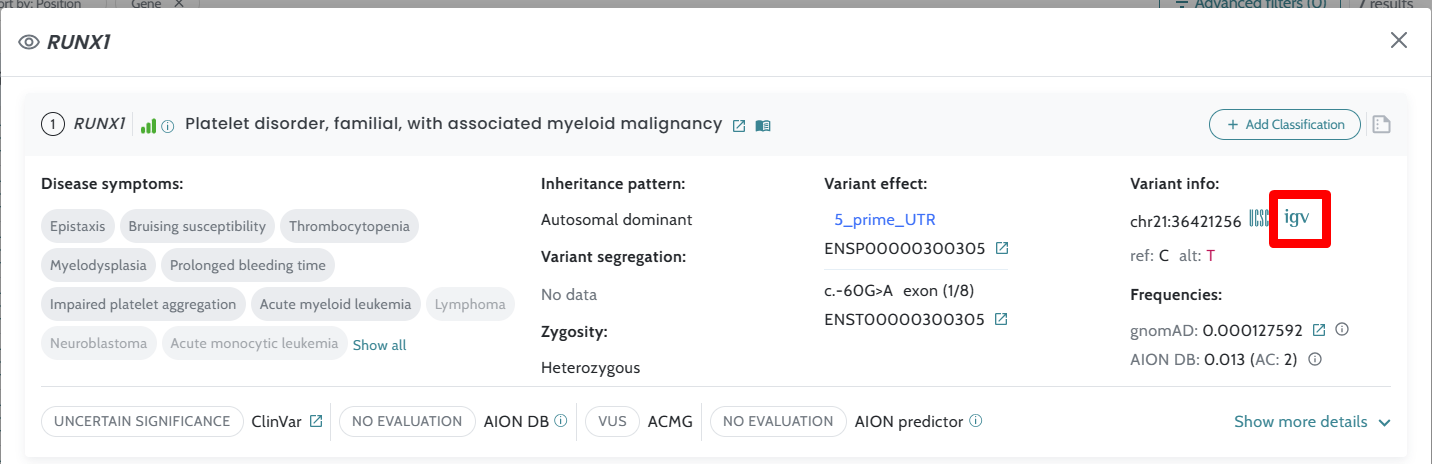
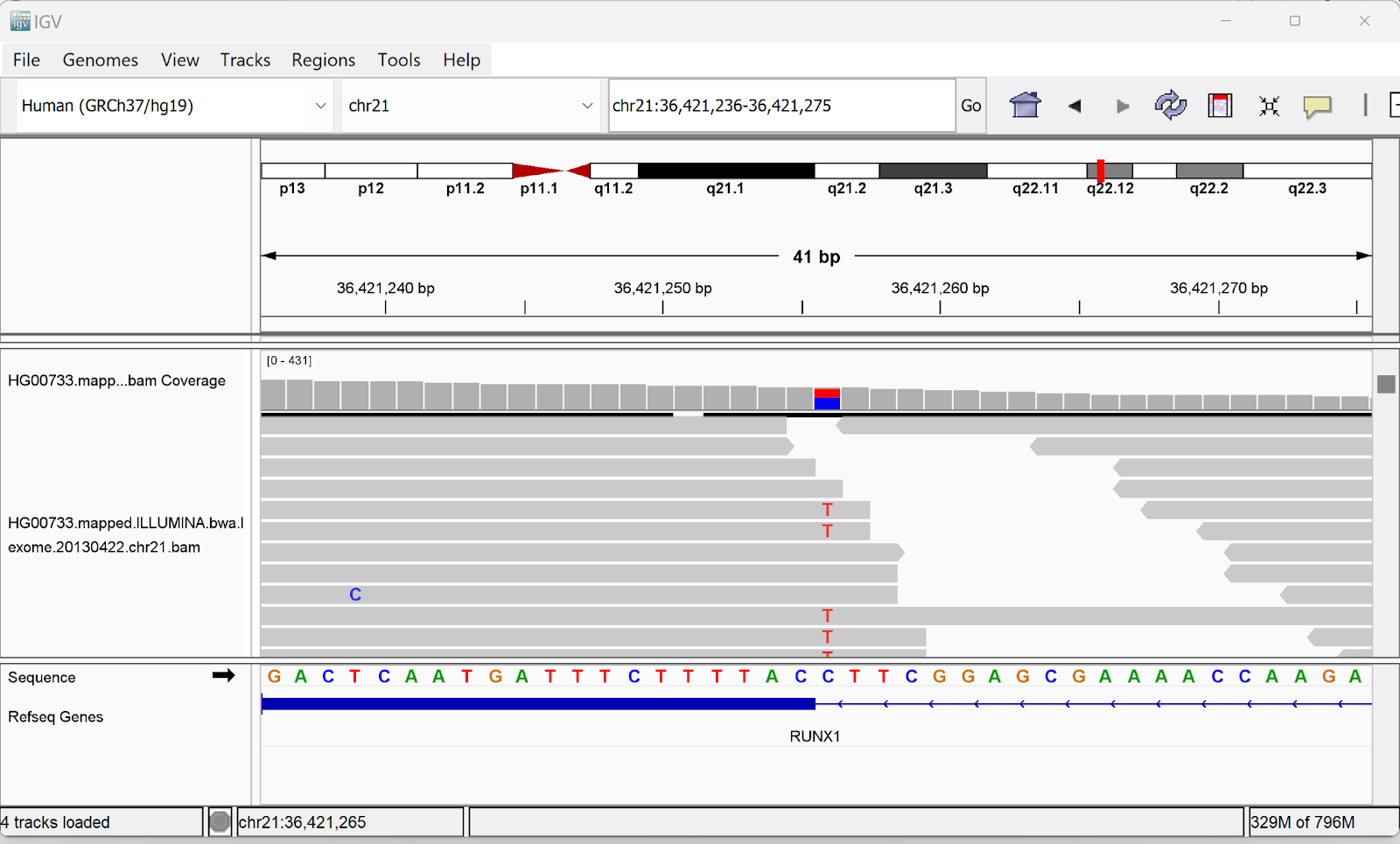
ℹ️ Find further visual support in the following clickable flow: Variant visualization with IGV
Each variant card can be expanded by selecting “Show more details” to view additional information about variant annotations, family info, literature and ACMG classification.
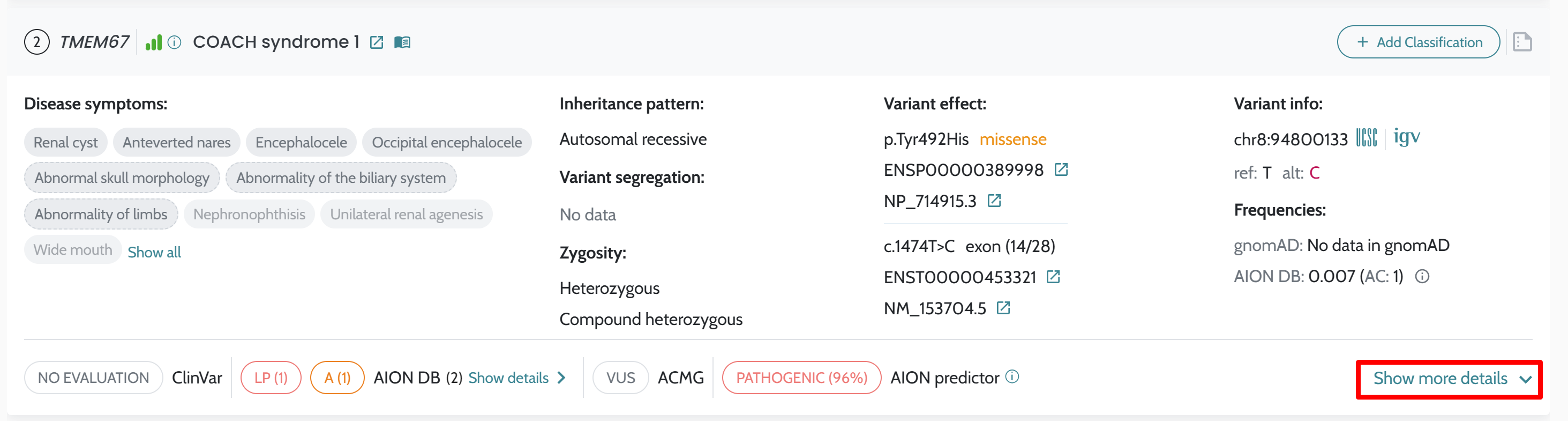
Click on Show more details > Annotations for each variant to see the following groups of selected annotations for each variant, related to Frequency, Evolutionary predictions, Splice and NMD predictions and Protein predictions
By clicking on the annotations, a glossary will appear explaining each annotation in detail.
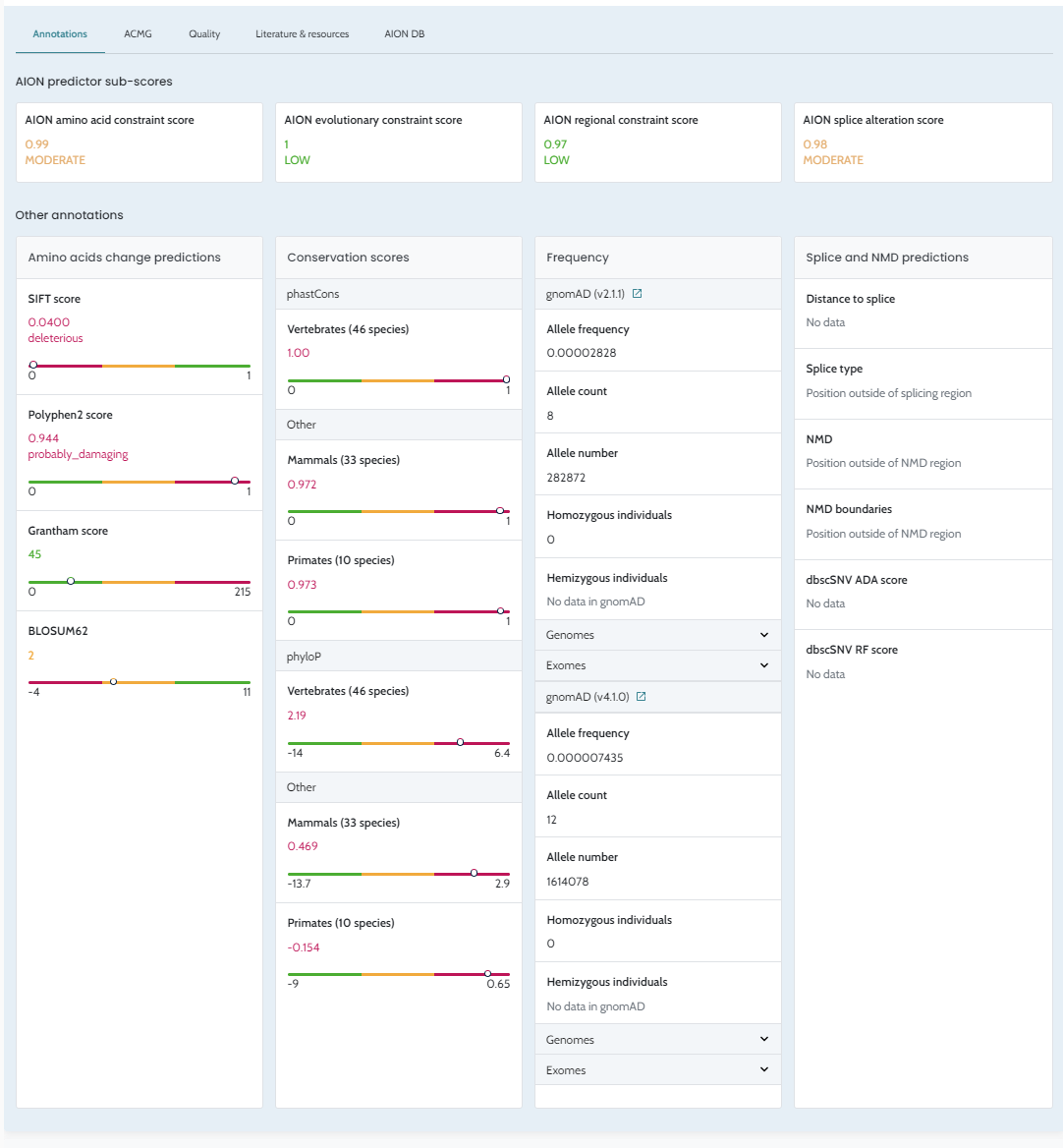
AION amino acid constraint: Indicates the predicted biochemical impact on the protein due to the amino acid change by variant. Values can be NA, High, Moderate or Low. The percentage indicates the confidence in the prediction considering available evidence. Values are calculated using a proprietary machine learning-based model.
AION evolutionary constraint: Indicates prediction of evolutionary constraint in the genomic region surrounding the variant. Values are calculated using a proprietary machine learning-based model. Values can be High, Moderate or Low. The percentage indicates the confidence in the prediction considering available evidence. Values are calculated using a proprietary machine learning-based model.
AION regional constraint: Predicts variation constraint in the genomic region surrounding the variant (as observed in healthy population individuals). Values can be High, Moderate or Low. The percentage indicates the confidence in the prediction considering available evidence. Values are calculated using a proprietary machine learning-based model.
AION splice alteration: Indicates prediction of likelihood of mRNA alterations caused by the variant. Values can be High, Moderate or Low. The percentage indicates the confidence in the prediction considering available evidence. Values are calculated using a proprietary machine learning-based model.
SIFT score: Values closer to 0 (range from 0 to 1) indicate deleteriousness. Predicts the effect of an amino acid substitution based on sequence homology and amino acid physical properties. If the score value is <= 0.05, the amino acid substitution will be predicted as “deleterious” and if the score is > 0.05, it will be predicted as “tolerated”. Values can be deleterious low confidence, tolerated low confidence, tolerated or deleterious. Values may be blank if the variant is not a missense change.
Polyphen2 score: Values closer to 0 (range from 0 to 1) indicate higher likelihood of pathogenicity. Predicts the effect of the possible impact of an amino acid substitution on the structure and function of protein. If the score is greater than 0.908, it will predict a “Probably Damaging” effect; if the score is greater than 0.446 and less than or equal to 0.908, it will predict a “Possibly Damaging” effect; and less than or equal to 0.446 will predict a “Benign” effect. Values can be blank if the variant is not a missense change, or if the value is missing from the annotation source.
Grantham score: Higher values indicate higher likelihood of pathogenicity. Informs on the evolutionary distance between two amino acids. Values range between 0 and 215. Values can indicate a conservative (0-50), moderately conservative (51-100), moderately radical (101-150), or a radical (≥151) change.
BLOSUM62: Lower values indicate higher constraint for an amino acid substitution and hence a higher likelihood of pathogenicity. It is a logarithm-of-odds score based on alignments between evolutionarily divergent protein sequences, providing a measure of the amino acid substitution frequency through evolution (i.e. indirect measure for specific amino acid substitutions constraint across evolution).
phastCons vertebrates (46 species): Values closer to 1 indicate higher conservation (values range from 0 to 1). Estimates the probability that each nucleotide belongs to an evolutionary conserved element in 46 vertebrates, based on multiple sequence alignment. PhastCons values consider not only each individual alignment column, but also its flanking columns. These values are sensitive to “runs” of conserved sites, and are therefore effective for picking out conserved elements.
phastCons mammals (33 species): Values closer to 1 indicate higher conservation (values range from 0 to 1). Estimates the probability that each nucleotide belongs to an evolutionary conserved element in 33 placental mammals, based on multiple sequence alignment. PhastCons values consider not only each individual alignment column, but also its flanking columns. These values are sensitive to “runs” of conserved sites, and are therefore effective for picking out conserved elements.
phastCons primates (10 species): Values closer to 1 indicate higher conservation (values range from 0 to 1). Estimates the probability that each nucleotide belongs to an evolutionary conserved element in 10 primates, based on multiple sequence alignment. PhastCons values consider not only each individual alignment column, but also its flanking columns. These values are sensitive to “runs” of conserved sites, and are therefore effective for picking out conserved elements.
phyloP vertebrates (46 species): Positive scores indicate a conserved region and so higher conservation constraint, while negative scores indicate accelerated evolution (scores ranging from -14 to 6.4). Specifically, it indicates evolutionary conservation at individual alignment sites for nucleotide/s changes in 46 vertebrates. PhyloP is specially appropriate for evaluating signatures of selection at particular nucleotides or classes of nucleotides (e.g., third codon positions, or first positions of miRNA target sites).
phyloP mammals (33 species): Positive scores indicate a conserved region and so higher conservation constraint, while negative scores indicate accelerated evolution (scores ranging from -13.7 to 2.9). Specifically, it indicates evolutionary conservation at individual alignment sites for nucleotide/s changes in 33 placental mammals. PhyloP is specially appropriate for evaluating signatures of selection at particular nucleotides or classes of nucleotides (e.g., third codon positions, or first positions of miRNA target sites).
phyloP primates (10 species): Positive scores indicate a conserved region and so higher conservation constraint, while negative scores indicate accelerated evolution (scores ranging from -9 to 0.65). Specifically, it indicates evolutionary conservation at individual alignment sites for nucleotide/s changes in 10 primates. PhyloP is specially appropriate for evaluating signatures of selection at particular nucleotides or classes of nucleotides (e.g., third codon positions, or first positions of miRNA target sites).
Allele frequency indicates alternate allele frequency for the variant of interest. This value is also displayed in the main variant card (variants can be filtered based on it).
Allele count indicates alternate allele count for the variant of interest.
Homozygous individuals indicate the total number of individuals homozygous for the alternate allele in the variant of interest.
Hemizygous individuals indicate the
total number of individuals hemizygous for the alternate allele
in the variant of interest. Notice only variants in sex
chromosomes can have a value in this entry.
⚠️ Notice that gnomAD frequency data include gnomAD v4.1.0 statistics. GnomAD v4.1.0 native data is aligned to GRCh38. For hg19 the values in gnomAD v4.1.0 are a liftover.
Distance to splice indicates distance to closest splice site in basepairs (1 to 10).
Splice type indicates the nature of the closest splice site (only shown for variants <10bp away from a splice site). Values can be blank (when the variant is not located in a donor or acceptor site), donor (GT) or acceptor (AG).
NMD indicates if the variant is predicted to escape nonsense mediated decay (NMD).
NMD boundaries NMD boundaries is a surveillance pathway that eliminates mRNA transcripts containing premature stop codons. A variant is predicted to escape NMD if it falls within any of the following regions, computed per transcript:
All regions include ±2 bp at intron-adjacent boundaries to capture splice-site variants (negative splice sites). NMD boundaries are computed only for transcripts with ≥2 coding exons, except for single_exon. Non-coding exons are discarded.
chr1:11822-11924(strand=1), the variant falls within an NMD escape region on the positive strand. The boundaries chr19:44677055 and chr19:44681836 represent the start and end of the region. Depending on the region label: for first_100bp, the start is ~52 bp upstream of the first coding exon and the end is 50 bp into it; for before-last-exon, the start is 55 bp upstream of the last exon–exon junction and the end is 2 bp into the downstream intron; for last-exon, the start and end flank the terminal coding exon by 2 bp.
chr2:14360-14831(strand=-1), the variant falls within an NMD escape region on the negative strand. Coordinates are always reported in genomic (not transcript) orientation, so the interpretation is mirrored: for first_100bp, the end is ~52 bp past the 5′ boundary of the first coding exon (in transcript coordinates) and the start is 50 bp into the exon; for before-last-exon, the start is 2 bp into the upstream intron and the end is 55 bp into the penultimate exon from its 3′ junction side; for last-exon, the start and end flank the terminal exon by 2 bp.
dbscSNV ADA score: ** Prediction score based on AdaBoost, an ensemble machine learning algorithm. Ranges 0 to 1, indicating the predicted probability that the variant will affect splicing. dbscSNV developers suggest a cutoff for a binary prediction (affecting splicing vs not affecting splicing) of 0.6.
dbscSNC RF score: ** Prediction score based on random forest, an ensemble machine learning algorithm. Ranges 0 to 1, indicating the predicted probability that the variant will affect splicing. dbscSNV developers suggest a cutoff for a binary prediction (affecting splicing vs not affecting splicing) of 0.6.
AION implements automated variant classification following the ACMG/AMP 2015 variant classification standards and ClinGen’s recommendations. The classification is computed using a points-based scoring system that evaluates a set of evidence criteria — known as ACMG rules — across multiple biological dimensions. Each rule contributes a score reflecting the strength of evidence for or against pathogenicity, and the sum of all applicable scores determines the final classification.
AION automates 23 of the standard ACMG/AMP rules. Three rules (PP4, BP2, BP5) are not currently automated and can be applied manually by the user. PP5 and BP6 have been deprecated following updated ClinGen recommendations (Biesecker et al., 2018) and are therefore not implemented.
Each rule is assigned a strength level, which translates into a numerical score:
| Strength | Direction | Points |
|---|---|---|
| Very Strong | Pathogenic | +8 |
| Strong | Pathogenic | +4 |
| Moderate | Pathogenic | +2 |
| Supporting | Pathogenic | +1 |
| Supporting | Benign | −1 |
| Strong | Benign | −4 |
| Standalone | Benign | −8 |
Some rules are applied at a fixed strength; others are applied at variable strength depending on the quality or weight of the supporting evidence (for example, the strength of a rule may be adjusted based on the review status of a ClinVar entry or the output of a computational predictor).
AION applies rules automatically based on the information available in the submitted VCF files and associated clinical data (e.g. HPO terms). Because some criteria require information that cannot be derived computationally — such as functional study results, detailed family history, or expert phenotypic assessment — some variants may initially receive a VUS classification pending manual review.
| Rule | Full name | Description |
|---|---|---|
| PVS1 | Loss-of-function variant | Frameshift, nonsense, canonical splice site (±1/2), and start-loss variants. Strength is determined by a structured decision process considering transcript relevance, NMD likelihood, and protein impact. See detail below. |
| PS1 | Same amino acid / splice-site change | Different nucleotide at the same position causing the same amino acid change or disrupting the same splice site as a known pathogenic ClinVar variant. Strength varies with ClinVar review status. |
| PS2 | Confirmed de novo variant | Variant arose de novo with both parentage confirmed through testing. |
| PS3 | Functional evidence of pathogenicity | ClinVar records a functionally abnormal effect for the variant. Can be upgraded manually following the BS3/PS3 decision tree. |
| PS4 | Variant prevalence in affected individuals | Heterozygous variant in an autosomal dominant context with significant phenotypic overlap and rarity in population databases. |
| PM1 | Mutational hotspot or critical functional domain | Variant falls within a functionally important domain or region enriched with pathogenic variants and absent benign variation, or has significant regional missense constraint. |
| PM2 | Absent or extremely rare in population databases | Variant frequency falls below inheritance-appropriate thresholds in large population databases. |
| PM3 | In trans with pathogenic variant (recessive) | In AR/XL disorders: variant found in trans with a known pathogenic variant (compound heterozygosity), or patient is homozygous/hemizygous with significant phenotypic overlap. |
| PM4 | Protein length change | In-frame insertions/deletions outside repetitive regions, stop-loss variants, and LoF variants in genes where LoF is not the primary disease mechanism. |
| PM5 | Different missense at disease-relevant codon | Different amino acid substitution at the same codon reported as pathogenic or likely pathogenic in ClinVar. Strength depends on ClinVar review status. |
| PM6 | Assumed de novo (unconfirmed parentage) | Variant absent from parents without formal parentage confirmation. |
| PP1 | Co-segregation with disease | Variant co-segregates with disease in one or more affected relatives. |
| PP2 | Missense in gene intolerant of missense variation | Missense variant in a gene where missense is a well-established pathogenic mechanism and benign missense variation is rare. |
| PP3 | Computational evidence of pathogenicity | Computational predictors support a damaging effect. A validated pathogenicity predictor is used for missense variants; a splice impact predictor for splice variants. Strength varies by score. |
Applies to frameshift variants, nonsense variants, canonical splice site variants (±1/2), and start-loss variants. The strength is not fixed: it is determined by working through a structured decision process that considers whether the transcript is biologically relevant, whether the aberrant mRNA is likely to be degraded by the cell (nonsense-mediated decay), how much of the protein is affected, and the predicted impact on splicing etc.. The strength can range from Supporting to Very Strong. PVS1 application is additionally capped by a gene-level assessment of disease association and whether loss-of-function is a well-established disease mechanism for that gene.
The decision logic follows the structured classification framework described in Abou Tayoun et al. (2018). Each branch of this framework is assigned a short code (e.g. NF1, SS3, IC2) that AION surfaces in the UI alongside the PVS1 result, so users can trace exactly which pathway was taken to trigger — or downgrade — the criterion.

| Rule | Full name | Description |
|---|---|---|
| BA1 | High allele frequency | Variant is common in large population databases. Standalone criterion — sufficient alone to classify a variant as Benign. |
| BS1 | Frequency exceeds disease threshold | Variant frequency exceeds what would be expected given the prevalence of the associated disease. |
| BS2 | Observed in healthy adults (early-onset disease) | Variant observed in sufficient unaffected adults in population databases, for a disease expected to manifest in early life. |
| BS3 | Functional evidence of no pathogenic effect | ClinVar records a functionally normal effect for the variant. Can be upgraded manually. |
| BS4 | Lack of segregation with disease | Variant absent in an affected family member (direct non-segregation), or arose de novo in an unaffected family member. |
| BP1 | Missense where LoF is primary mechanism | Missense variant in a gene where the primary pathogenic mechanism is loss-of-function, making a missense change less likely to be causative. |
| BP3 | In-frame indel in repetitive region | In-frame insertion/deletion in a repeat or low-complexity region without known functional importance. |
| BP4 | Computational evidence of benign effect | Computational predictors support a benign effect. Strength varies by predicted score. |
| BP7 | Synonymous with no predicted splice impact | Synonymous variant with no predicted splice effect and low evolutionary conservation. |
Some ACMG/AMP evidence codes are not annotated automatically by AION because they require information which is challenging to process computationally, or not present in the VCF file.
| Rule | Full name | Description |
|---|---|---|
| PP4 | Highly specific phenotype | Patient’s phenotype or family history is highly specific for a disease with a single genetic etiology. |
| BP2 | In trans / in cis with pathogenic variant | Observed in trans with a pathogenic variant for a fully penetrant dominant gene/disorder, or observed in cis with a pathogenic variant in any inheritance pattern. |
| BP5 | Alternate molecular basis | Variant found in a case with an alternate molecular basis for disease. |
ℹ️ PP5 and BP6 were discontinued based on the recommendations by ClinGen (Biesecker et al., 2018)
The sum of all applicable rule scores produces a total score that determines the final classification:
| Total Score | Classification |
|---|---|
| ≥ 10 | Pathogenic |
| 6 – 9 | Likely Pathogenic |
| 5 | VUS – Hot |
| 4 | VUS – Warm |
| 3 | VUS – Tepid |
| 2 | VUS – Cool |
| 1 | VUS – Cold |
| 0 | VUS – Ice Cold |
| −1 to −6 | Likely Benign |
| ≤ −7 | Benign |
Variants of Uncertain Significance (VUS) are sub-classified on a scale from “Ice Cold” to “Hot” to provide clinicians with additional granularity about the weight of available evidence, even when it is insufficient to reach a definitive classification.
When two rules would produce contradictory evidence, the higher-priority rule takes precedence and the lower-priority rule is suppressed automatically. The complete set of enforced mutual exclusions is as follows (the first rule listed takes priority):
| If this rule fires… | …this rule is suppressed |
|---|---|
| BA1 | BS1 |
| PM1 | BP3 |
| PM2_supp | BS1 |
| PM4 | PP3 |
| PS2 | PM6 |
| PP2 | BP1 |
| PVS1 | BP4 |
| PVS1 | PM4 |
| PVS1 | PP2 |
| PVS1 | PP3 |
| PVS1 | PM1 |
| BP7 | BP4 |
| BA1 | BA1 stands alone as benign, regardless of any other triggered rules |
For the full ACMG/AMP classification framework, refer to the original publication: Richards et al., Genetics in Medicine, 2015.
AMP evidence codes annotated by AION are displayed in the ACMG tab. Criteria are organised by several properties, such as Segregation, Disorder, Effect, etc. “Selected by AI” means that the criterion is automatically applied by AION.
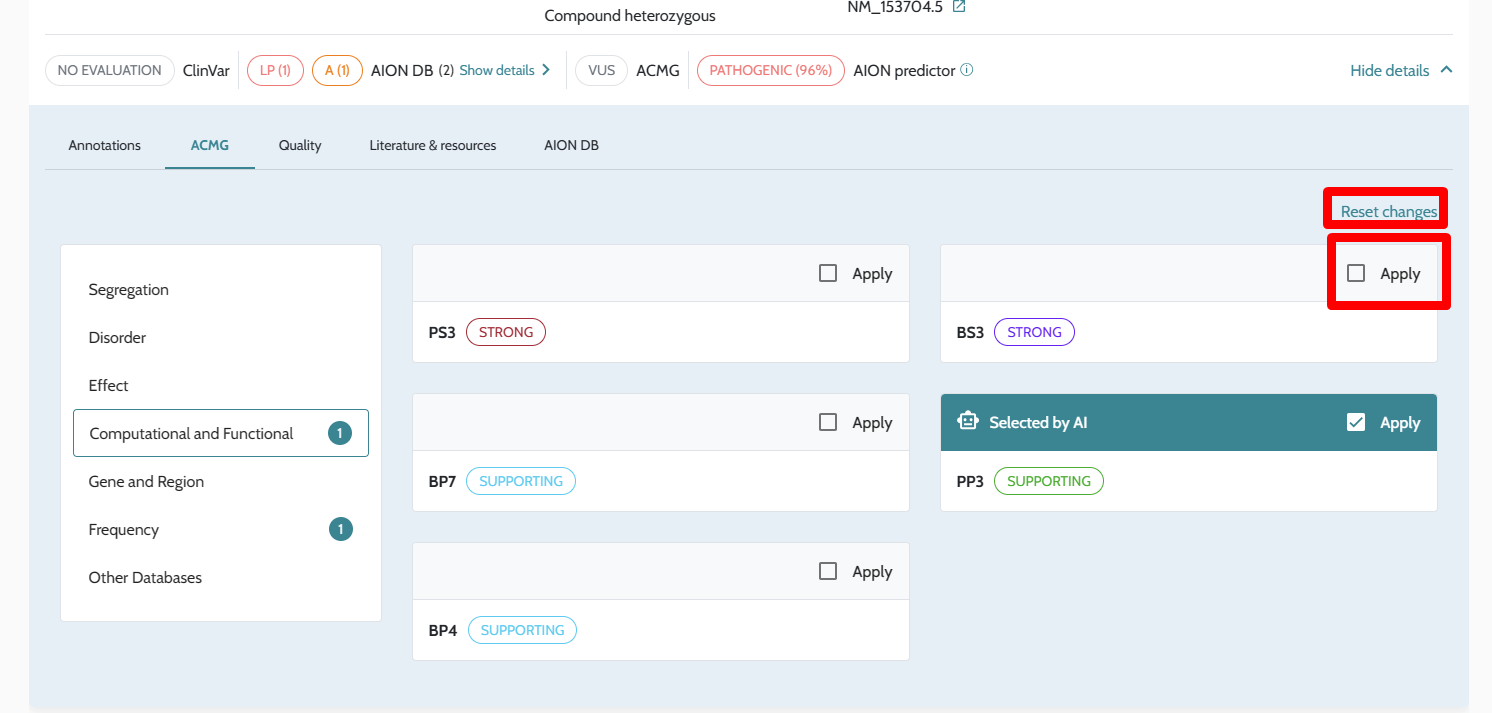
In order to improve variant classification, the user can manually apply or remove any evidence code, based on the evidence they have for a particular variant in a patient by selecting or unselecting “Apply”. The new ACMG/AMP classification will be calculated on the fly. By selecting Reset changes, user-selected changes can be reset and the automatically applied criteria shown again.
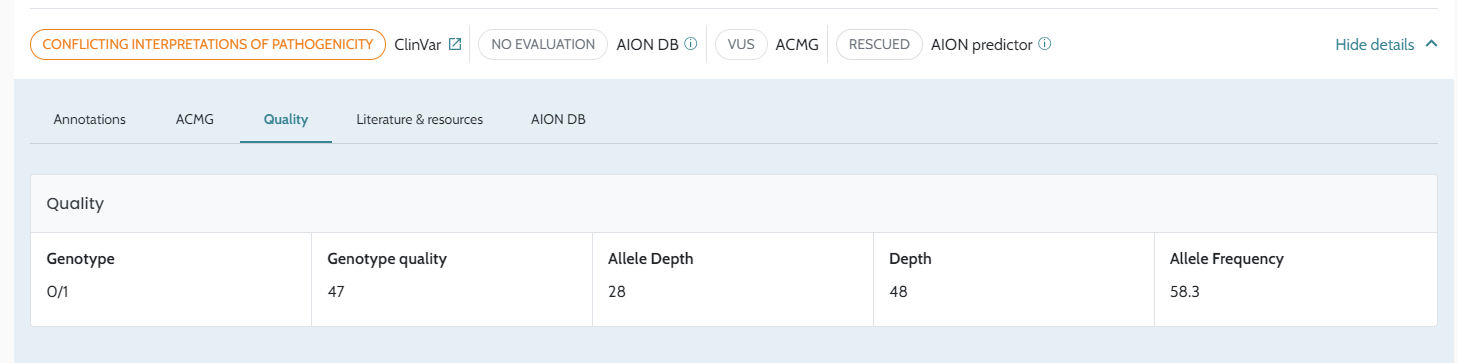
Genotype (GT)
Genotype quality (GQ) represents the Phred-scaled confidence that the genotype assignment (GT) is correct.
Depth (DP) refers to the filtered depth (the number of filtered reads that support each of the reported alleles). Only reads that passed the variant caller’s filters are included in this number. However, unlike the AD calculation, uninformative reads are included in DP.
Allele depth (AD) is the unfiltered allele depth, i.e. the number of reads that support each of the reported alleles. Allele depths (ref, alt) are separated by commas. All reads at the position (including reads that did not pass the variant caller’s filters) are included in this number, except reads that were considered uninformative. Reads are considered uninformative when they do not provide enough statistical evidence to support one allele over another.
Variant Allele frequency (VAF) is the percentage of sequence reads observed matching a specific DNA variant (alternative reads) divided by the overall coverage at that locus.
This section currently provides direct links to:
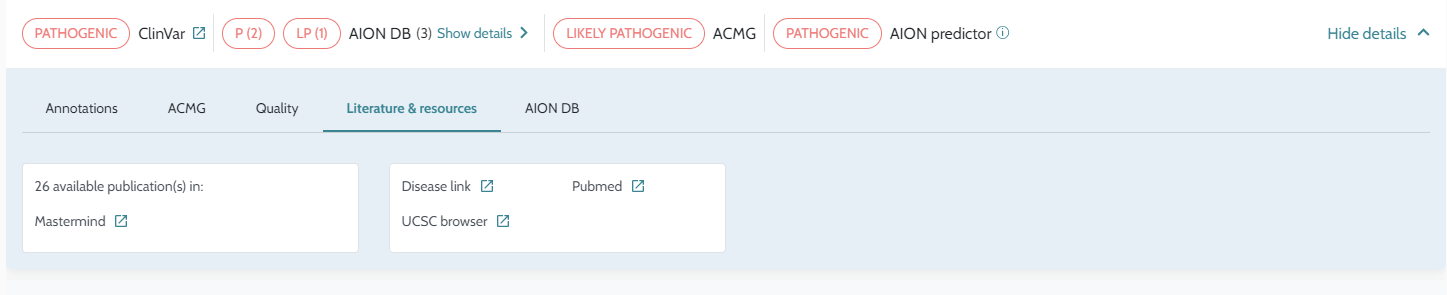
ℹ️ Find further visual support in the following clickable flow: Results Review - Small Variants
ClinVar classifications are integrated into AION’s variant interpretation alongside other evidence sources — including ACMG/AMP criteria, population frequency, and computational predictors — to contribute to the overall pathogenicity assessment of each variant.
The ClinVar value shown on a variant card comes from ClinVar Nostos, AION’s internal processing layer that consolidates potentially conflicting ClinVar submissions from multiple submitters into a single conservative classification. For a full description of how this consolidation is computed, see ClinVar Nostos Classification Logic.
During variant processing, AION’s quality control pipeline assigns filter tags to variants that show signs of being artefactual or of insufficient quality for reliable interpretation. Variants carrying filter tags may be excluded from the prioritised results or ranked lower, depending on the tag type and severity.
Understanding these tags helps you interpret why a variant may not appear in the Relevant Variants or AION Clues lists, and whether it warrants manual review via the All Variants view.
Two output columns directly reflect the outcome of quality filtering for downstream use:
n_filter_tags: The number of
filter tags assigned to a variant. A value of 0
means the variant passed all quality checks and is eligible to
appear in AION Clues. Any value greater than 0
indicates the variant has been flagged and will not be included
in the prioritised results.is_smoking_gun: Present in the
prioritiser output file. A value of 1 indicates the
variant has been selected as an AION Smoking
Gun; 0 means it has not. This column can
be used programmatically to filter or highlight the strongest
diagnostic candidates.For questions about specific filter tag combinations or edge cases not covered here, contact support@nostos-genomics.com.
ℹ️ Filter tags are visible in the downloadable annotation
file (TSV) for small variants in the filter_tag
column. Reviewing this column can help diagnose why specific
variants are absent from prioritised results.
⚠️ Variants flagged by filter tags are not deleted — they remain in the annotation output and are accessible via the Manual Filtering view. If you believe a flagged variant is clinically relevant, you can review it there and apply your own interpretation.
Please see the dedicated pages:
Additionally to the AION results tab, all annotated variants are available for analysis on Manual variant filtering - Small variant view.
⚠️ AION annotates all variants of the VCF file that are in
the supported regions and has VCF
FILTER value PASS or “.”
Be aware that depending on your secondary analysis configuration
this might mean also variants of low quality can be visualised
and available for analysis through the manual filtering view!
You can set filters for variant quality directly in the column
filters, or utilising advanced
filters.
The goal of AION is to provide high quality automated analysis so that users don’t have to use manual filtering of variants. The AION results tab is the best place to find relevant variants fast. It shortlists the variants that are relevant from a molecular and a clinical perspective, drastically improving the interpretation efficacy. For more details on the AION results (Smoking guns and AION Clues) visit the AION results page.
However, in those cases where additional information is needed or it is simply desirable to double check whether no other relevant variants were present in a given region, it is possible to use the manual filtering. This could happen, for instance, when the case phenotype is not detailed enough to drive accurate clinical prioritization or when there is strong suspicion that the causative variant may be in regions not supported by the automatic pipeline (see details here).
The manual filtering - small variants tab consists of:
A filterable table with all annotated variants allowing one by one filtering in the column filters and sorting of results
An advanced filters filter functionality ( see page for creating, editing and applying advanced cascading filter logic with just a few clicks , see more details Advanced filters
Some variants may be listed multiple times if their position is a region of overlap between two genes. In that situation, the variant would be shown twice, one for each associated disease.
Each variant in the table has a detailed view informing the user about its clinical relevance, linking it to one or more diseases (when available) as well as allowing the user to add the specific variant-disease pair to the report.
CNV results are currently visualised on Manual filtering - CNVs tab.
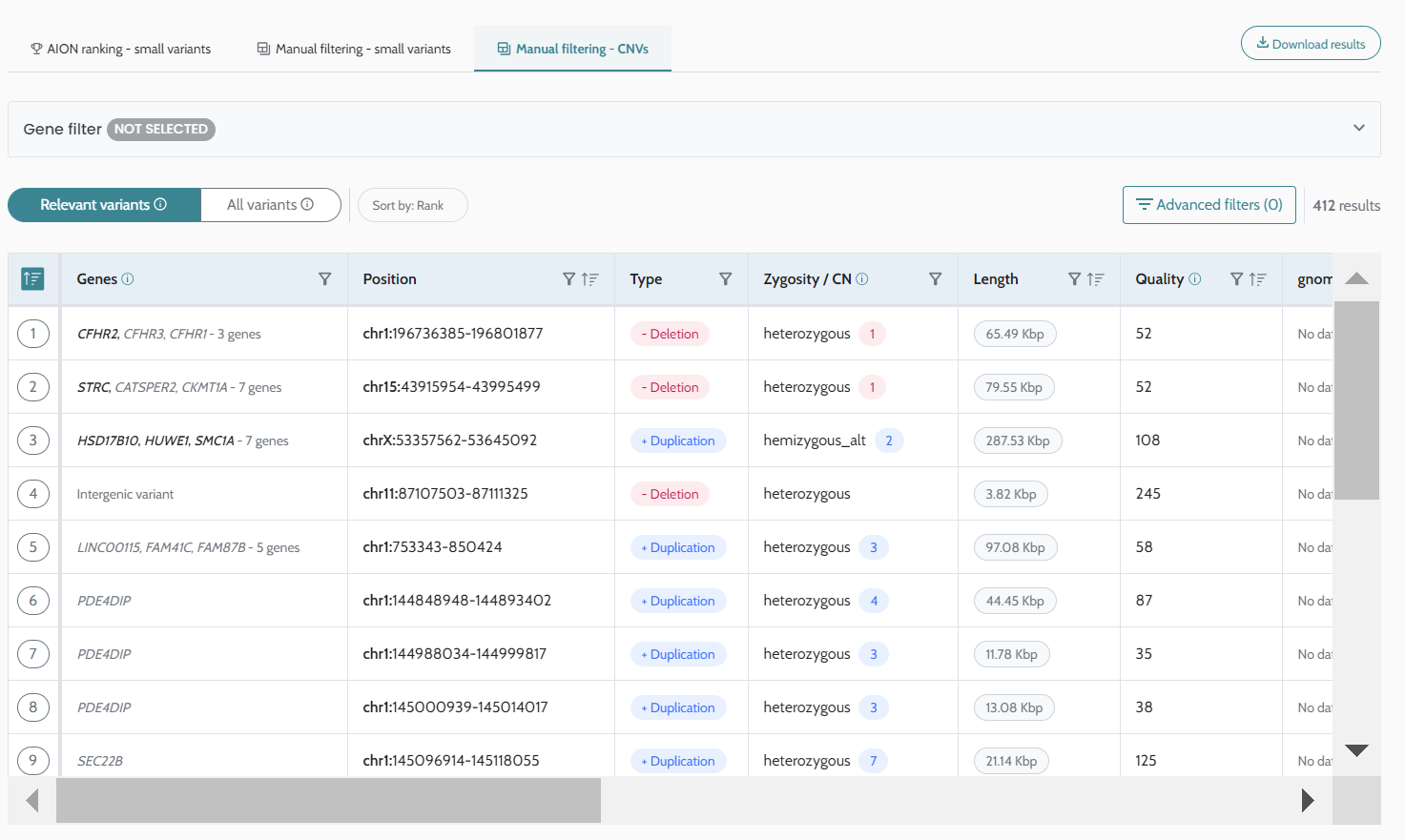
ℹ️ AION shows in bold the genes associated to diseases in the ranked variant list. Notice that for CNVs the association to disease is shown only when the disease has associated HPO terms.
The manual filtering - CNVs tab consists of:
A filterable table with all CNV variants annotated by AION. By default the table visualises high quality variants only (see definition of high quality CNV variants here: (VCF format | CNV / SV variants VCFs) and is sorted by rank.
An “Advanced filters” filter functionality for creating, editing and applying advanced cascading filter logic with just a few clicks ,see more details Advanced filters
Each variant in the table has a detailed view to inform the user about its clinical relevance through annotations and diseases linked to genes affected by the variant.
Currently AION visualises and annotates all CNVs with the following information.
Basic variant information annotated
Type of CNV
Position
Zygosity and copy number (CN). The zygosity is deduced from the CN as follows:
Length
Quality
Position of the CNV in genome
Cytoband
Related genes
Filter status in AION
File the variant comes from Each associated gene is further annotated with;
Diseases linked to the gene and its
Inheritance pattern
Symptoms of the diseases and a full comparison to patient symptoms
Clingen HI/TS dosage sensitivity scores
pLI, LOEUF, pHaplo, pTriplo Association to known classified CNVs (pathogenic or benign) from multiple databases. The following sources are used:
Pathogenic sources;
Benign sources;
All variants have a link to the IGV desktop application to allow visual exploration of the genomic data. To be able to visualize the data you should have the IGV desktop application open with the corresponding reference genome and relevant BAM files. Additionally, port listening must be enabled and configured to the default port (60151) in the Advanced tab of the View > Preferences window (refer to IGV for more information).
 **
**
ℹ️ Find further visual support in the following clickable flow: Variant visualization with IGV
The Advanced filters allows you to create reusable filtering queries that can be applied to all your cases. Advanced filters allow you use complex logic to search for relevant variants by filtering on different quantities. Additionally, you can use advanced filters to define and store in silico gene panels. Since advanced filters can be shared across an institution, they can be used to enforce institutional best practices both on the filtering as well as on the in silico gene panels approaches.
Advanced filters may be created based on filters on individual quantities as well as based on other already defined advanced filters, allowing you to build complex filter logics. Advanced filters are available for all users and can be accessed on either small variant or CNV manual filtering pages.
When selecting Create new filter, the user is asked to give a name to the advanced filter. Manager users can also define whether it is a private or an organisational filter.
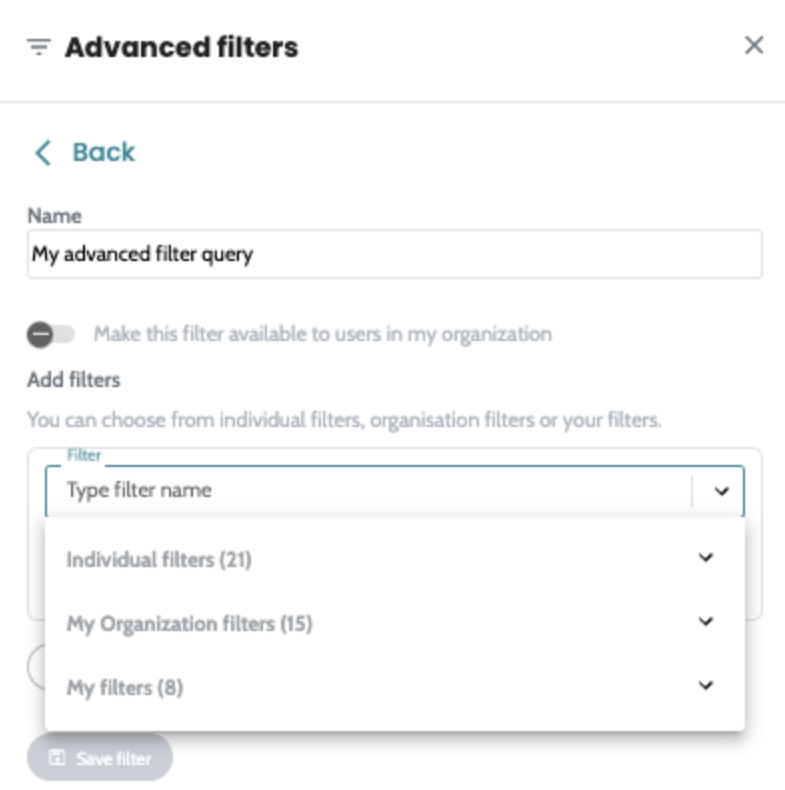
Then simply add steps to your advanced filter. A step may consist of one of the following three options:
Individual filters - Add an individual quantity to filter by and define the criteria to use. Note that individual filters may be specific to the variant type (Small variant, CNV) and there cannot be conflicting variant types in an advanced filter. So if you add a filter that only applies to CNV variants to an advanced filter, you cannot add one that only applies to small variants, nor apply that advanced filter in the small variant view.
My organisational filters - Select from previously created advanced filters shared within your organisation.
My filters - Select from your own previously defined advanced filters.
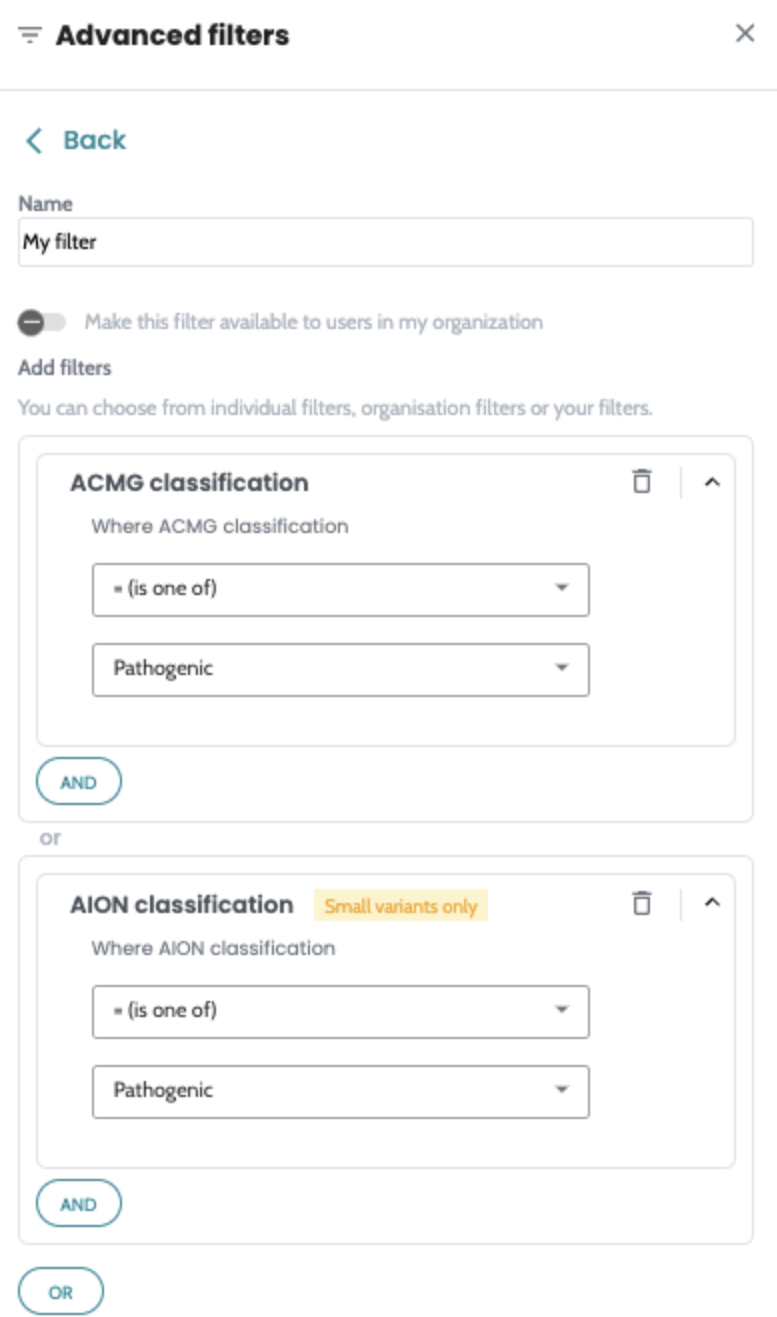
You can use different logic to chain steps together. Steps in
an advanced filter may be chained by either AND or
OR operators as seen in the image on the right.
Advanced filters enforces steps as concepts, so it is recommended that you define advanced filters for specific concepts and then create other advanced filters to chain those together. A practical example of this is the following logic:
(FilterConsequence) AND (FilterFrequency)
The best practice is to define an advanced filter for
FilterConsequence and another one for
FilterFrequency before chaining them together in a
third advanced filter. The advantage of following this pattern
is that any institutional update on the definition of
FilterConsequence or on
FilterFrequency would automatically be applied to
all advanced filters that includes both.
When applying advanced filters drawer you will see two types:
You can apply a single advanced filter or apply multiple
combining them with either AND or OR
operators. If more complex logic is needed to combine multiple
advanced filters, that can be easily done by creating another
advanced filter.
ℹ️ Find further visual support in the following clickable flow: Advanced Filtering - Cascading Filter sets
As part of the AION Database (AION DB), data submitted to AION automatically generate statistical insights from all the cases you submit. This statistical data is then annotated to new cases and can be utilised as additional source of insights when analysing the case.
The AION DB is a private database on a per customer level to store and generate insights. Any information stored is not shared with other accounts. The AION DB contains two types of data:
For both types of data, only small variants (SNPs and Indels) are currently supported. CNV variants and other will be supported in a future iteration of the functionality.
In this article you will find more information about the statistical data generated. The AION DB also stores variant classification data, please refer to that article for further information.
Access to the statistical insights of the AION Database is activated through our support team. If you don’t have it active, please get in touch with our support team at: support@nostos-genomics.com
You can leverage AION DB variant statistic and previous classifications for filtering variants in the manual filtering views through either column filters or advanced filters.
AION currently computes the following statistical data:
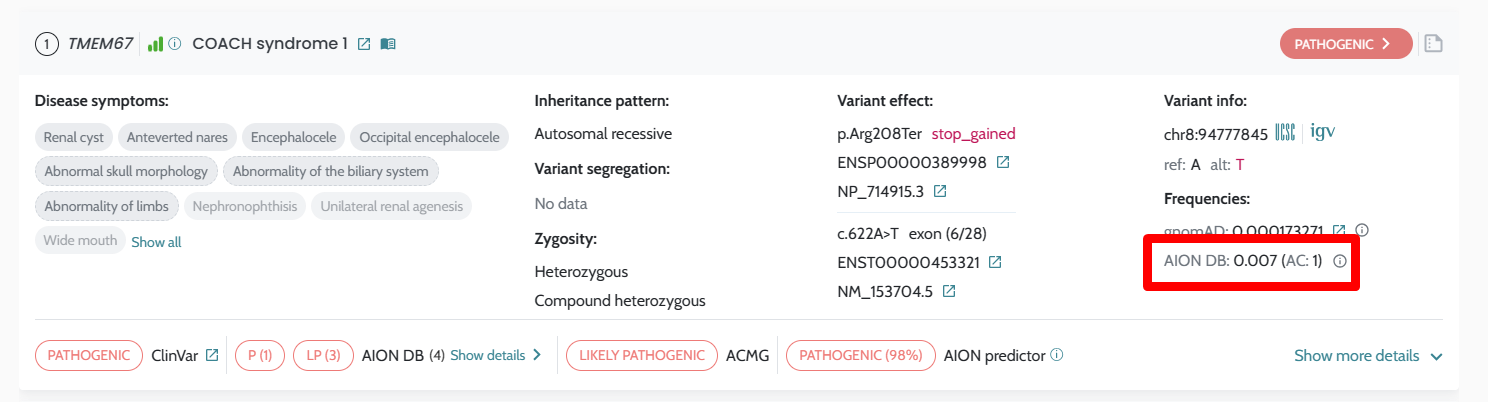
Each of these quantities are computed for the total population as well as for affectedness status (affected / not affected). This is intended to accommodate frequency data coming from control populations or from unaffected parents in duo/trio/etc cases.
Importantly, cases with different reference genomes are also kept at different databases.
AION displays the data in the AION DB in the AION ranking. The variants that are present in the AION DB are highlighted in the AION ranking showing the allele count of the variant in the AION DB, the population frequency and the past interpretations available.
If the variant is found in the internal variant database, a tab will be available containing the past interpretations along with how many cases contain this variant, the zygosity of the variant in those cases and the affectedness status.
Additionally, you can filter by this data in the manual filtering views.
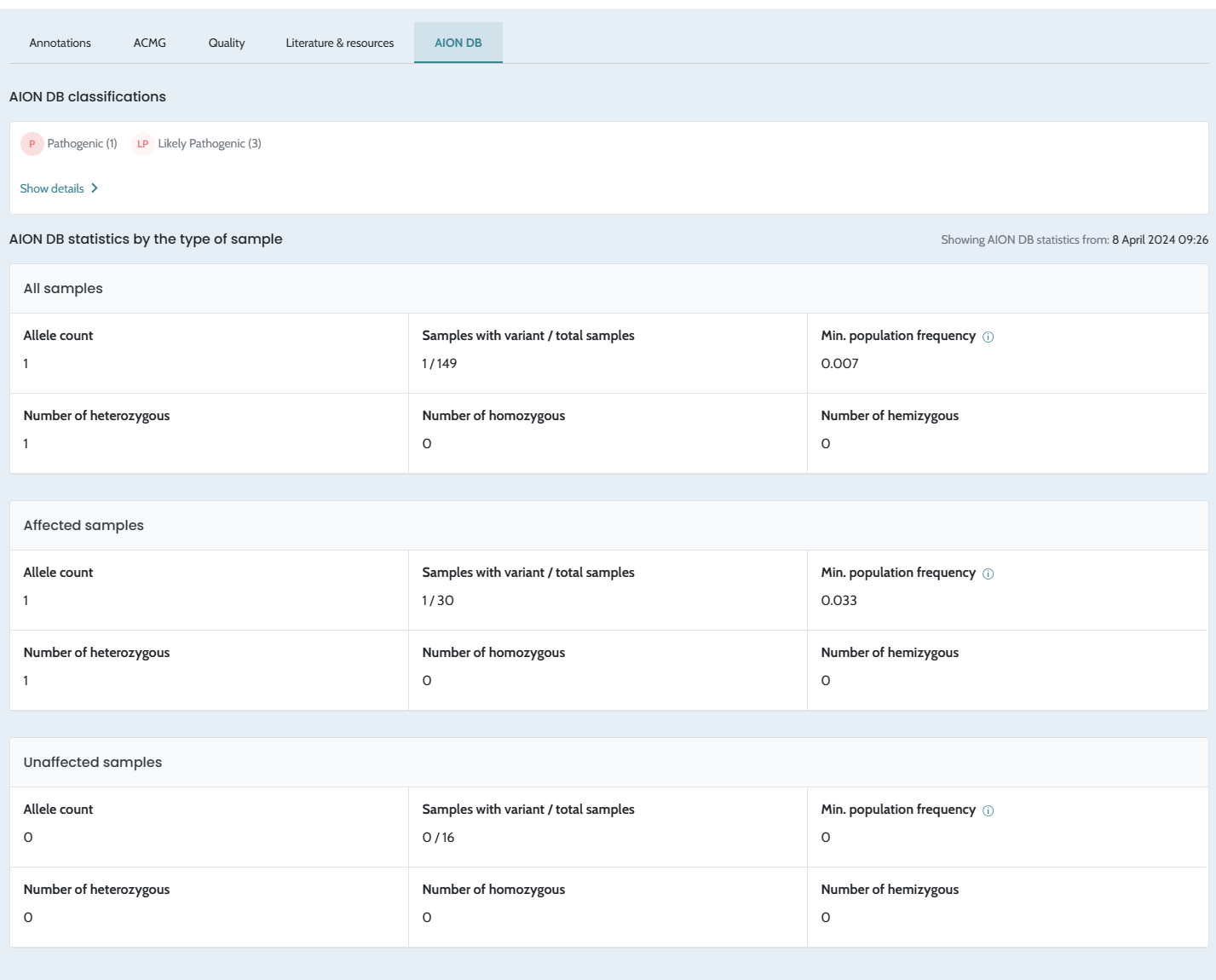
Cases that have more than 7 days since submission and have not been accessed during the last 24h become inactive. To be able to check the status of the case, you need to access the case to visualize this information. In order to activate the case, press the “Activate” button.
Case activation will take a few minutes (2-10mins) and will enable the ability to fiter by AION DB statistics in the manual filtering - small variants tab.
Upon the activation of the case you will be able to refresh the statistics as described in the Timing section below.
Variant statistics are annotated from the annotation-ready data from the AION DB as it is at submission time. The annotation data is refreshed every 30 minutes during business hours (6am-10pm CET), so new cases during these hours will be annotated with, at most, 30 minutes old data. You will always see a date and time of the statistics shown for each case.
The AION DB statistics data can be updated by pressing the “Refresh” button on the case header information. AION currently support updated for data analysed using GRCh37/hg19 reference genome:
The ranking is not affected by the data in the AION DB,
however, data is clearly displayed for the consideration of the
user.
The population frequency is effectively a minimal population frequency because there is no information on the sequenced regions in the VCF files used as input. It can be that a specific VCF file does not contain a specific variant because that variant is not present in the sequenced individual or because the region where the variant is located was not sequenced. This cannot be solved using data from VCF, so we display the calculated population frequency noting this important remark.
The current case has no special treatment in the AION DB and will be considered towards the statistics. However, given the timing of when new data is annotated onto cases, at first the current case will not count towards the statistics. It will only do so if statistics are manually refreshed or if the case is relaunched and the annotation-ready data in the AION DB has already been refreshed.
We check for duplicates in the AION DB, so if you submit the same VCF file twice, it will not be added again. However, this wouldn’t recognise a re-sequencing of the same person in a new VCF file.
ℹ️ Find further visual support in the following clickable flow: AION DB - Statistics
See AION Database - onboarding
As part of the AION Database (AION DB), classifications recorded by users in AION are automatically stored into AION DB and then available for annotation when analysing the same variant again in another case. This data is then annotated to new cases and can be utilised as additional source of insights when analysing the case.
The AION DB is a private database on a per customer level to store and generate insights. Any information stored is not shared with other accounts. The AION DB contains two types of data:
For both types of data, only small variants (SNPs and Indels) are currently supported. CNV variants and other will be supported in a future iteration of the functionality.
In this article you will find more information about the variant classification options. The AION DB also stores variant statistics data, please refer to that article for further information (see here).
Previous variant classifications are key to generate structured institutional knowledge. Through this method, you can leverage and review previous decisions. Each variant classification action requires a comment by the user to justify the decision.
For small variants variants, AION DB classifications are
annotated per nucleotide change on new variants. Therefore,
means, when analysing a variant in AION, you will see any
classification from the database for the same variant (defined
as: chr-pos-ref-alt). This matching is done
independently of zygosity, gene/transcript, disease and
inheritance mode.

Summary of the AION DB classifications are visualised in the footer of variant cards. More detailed information is visualised in the variant classification drawer, which can be opened by selecting Show details.
Previous classifications can also be used for variant filtering, allowing you to efficiently accumulate knowledge. You can leverage AION DB variant statistic and previous classifications for filtering variants in the manual filtering views through either column filters or advanced filters.
Recording final decisions of variant pathogenicity can be done through the variant classification flow in AION. Any small variant can be classified in AION, so the same flow applies to variants in the AION ranking and variants in manual filtering tab.
To start recording your decision, select “Add classification” button in the upper right corner of the variant card.
Selecting Add Classification opens a the variant classification drawer, where you can:
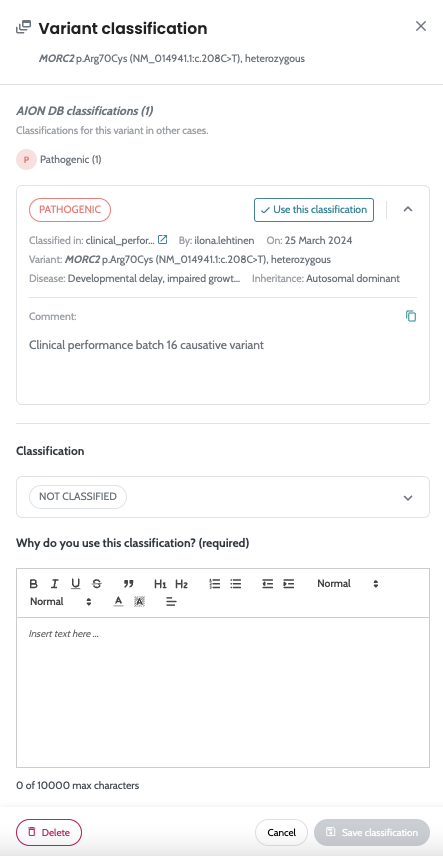
A classification of a variant can be edited at any time. Changes of the classification are logged, and visualised in the drawer.
The classifications that AION allows to give to a specific variant are: Pathogenic, Likely Pathogenic, VUS, VUS to Report, Likely Benign, Benign, Artifact and Other. However, we would discourage using “Other” since this classification as it is not specific category or related to the pathogenicity of the variant and can be difficult the interpretation of the variant in future by yourself or a team member.
⚠️ A classification can also be deleted. This process is irreversible and must be used with caution.
Note! Currently variant classification is not connected to reporting, and variants need to still be selected to reported separately. Please refer to page Generating a report for information related report generation.
ℹ️ Find further visual support in the following clickable flow: AION DB Manual Variant Classification
During onboarding, you may submit past samples and past classified variants to the AION DB in order to carry information previously generated at your institution.
For this, three resources need to be provided to the support team. The data submission is done through direct communication with the support team. Sample files are provided to facilitate the process.
The required resources, along with the required information are:
Classified variants: A TSV file containing classified
variants. It should contain the following data: chromosome,
position, ref, alt, zygosity, past interpretation, sample ID
(same as the VCF data files the the classified variant is also
in one of them), affected status, reference genome, original
classification/submission time and the person who originally
classified/submitted the variant.
The following is a detailed description of allowed values for each field of the TSV files:
VCF metadata file:
Classified variants file:
The manager component of AION is intended to be used mostly by clinical laboratory managers and requires admin permissions. You can access it by selecting your user name in the upper right corner → Manager.
Here you can:
Browse list of current users and their roles (manager, user, admin) and the date of creation of the user
Edit users by hovering over a user and selecting Edit. The following actions are supported: edit username, edit roles or deactivate/activate a user.
Create new user by selecting “Add a new user”
You can have access to the latest version of AION by making a request through the Nostos Genomics website.
Simply provide your name and last name, as well as a valid company email address (free email services are not allowed) and after approval you will receive an email invitation to AION variant interpretation platform. Follow the instructions and you will have an account to test the future of variant interpretation.
All users using an email account with the same domain will be
added to a same company account. 
In this section, we provide illustrative examples of how AION can help identify causative variants in patients.
To run these cases, simply follow these steps:
Then follow the usual results analysis procedure.
Case 1 is a patient affected by a very specific ophthalmological disease. A variant in CNGB1 was strongly prioritized in Rank #1 due to a very strong match between the candidate disease and patient’s phenotype, and also due to being an homozygous variant in a gene associated to autosomal recessive inheritance. Furthermore, this variant was also correctly prioritized thanks to its description in Clinvar, even if it was annotated as “Conflicting interpretations” (as some submitters annotated it as Pathogenic, others as Likely Pathogenic), showing how AION correctly leverages information from databases.
Sample VCF:
HPO terms:
Causative variant:
Case 2 is a patient affected by neurodevelopmental delay. A variant in KIF1A was prioritized in rank #1, not only due to phenotype matching between the patient and the candidate disease, but also due to AION’s ML score prediction of pathogenicity (91%). After performing variant segregation in the parents, this variant was shown to be de novo (absent from the parents), thus confirming its pathogenicity. Interestingly, this variant was later annotated as Likely Pathogenic in ClinVar due to description in other patients. This case illustrates how AION machine learning algorithms can perform correct predictions of pathogenicity in cases where the variant has not been previously described in databases!
Sample VCF:
HPO terms:
Causative variant:
Case 3 is a patient affected by a multisystemic disease including delayed bone growth. Very accurate phenotypic description enabled the identification of 2 previously undescribed variants in ADAMTSL2, which have later been confirmed as pathogenic . Interestingly, these 2 variants are in compound heterozygosis, exemplifying how AION is able to identify and prioritize compatible compound heterozygous variants, even when none of them have been previously described. Of note, these 2 variants were also prioritized on top ranks (#1 and #2) even if this trio case is run without parents. Nevertheless, running trio samples helps to shorten “diagnostic odyssey” because having segregation information prevents geneticists to perform additional tests (e.g. Sanger sequencing) to confirm variant cosegregation in parents.
Sample VCF:
HPO terms:
Causative variants:
Case 4 is a patient affected by a neurodevelopmental disorder with very characteristic facial features. In this case, a missense variant in NIPBL was strongly prioritized in Rank #1 due to a very strong match between the candidate disease and patient’s phenotype. This variant was shown to be de novo thanks to trio sequencing; nevertheless, when running this case as a singleton, this variant was also prioritized in rank #1, proving how AION is able to prioritize causative variants with little or no information from databases. While in the trio scenario prioritization would be driven by our implementation of ACMG criteria (tagging this variant as Pathogenic), in the singleton scenario we can see that prioritization is also driven by AION’s ML score prediction of pathogenicity (ML 96%), based on our in-house machine learning model and annotation from >100 different databases.
Sample VCF:
HPO terms:
Causative variant:
Case 5 is a patient affected by a severe immunological disorder. Partly thanks to accurate laboratory-related HPOs (e.g. Reduced granulocyte CD18 level), an homozygous loss-of-function variant in FERMT3 was strongly prioritized in Rank #1. This case exemplifies how important is an accurate phenotypic characterization, even of HPOs that are not related to visible physical symptoms.
Sample VCF:
HPO terms:
Causative variant:
Case 6 is a patient affected by a congenital gastrointestinal disorder. In this case, AION was decisive to provide a quick diagnosis to a newborn patient. By identifying these 2 variants in SLC9A3 (coding Na(+)/H(+) antiporter 3, NHE3 protein) in compound heterozygosis, described in ClinVar, in a case with a very strong phenotypic match with the candidate disease, a fast diagnosis of this case was possible, enabling a better managing of this patient since a very young age and genetic counselling.
Sample VCF:
HPO terms:
Causative variants:
Case 7 is a patient affected by nonsyndromic intellectual disability, speech impairments and motor delay, described in more detail this paper (SRR11604298). Patients data has been analysed with Nostos DRAGEN secondary analysis pipeline for small variants, structural variants (SVs) and copynumber variants (CNVs).
The causative variant, a large duplication in chrX has been strongly prioritised in Rank #1 thanks to correct ACMG classification powered by annotation of overlapping known pathogenic CNV in dbVar, as well as strong clinical overlap between the patient and the diseases linked to associated genes.
Sample VCFs:
HPO terms:
Causative variant:
ℹ️ Any serious incident that has occurred in relation to the device shall be reported to Nostos Genomics GmbH (see contact details below) and the competent authority of the EU Member State in which the user and/or the patient is established.
Nostos Genomics GmbH can be contacted through:
AION’s availability as a SaaS Infrastructure is 95% per month.
For further information regarding the treatment of personal and other types of data, please refer to the privacy policy (available here).
Additionally to the VCF format checklist in VCF format , these are things to consider to guarantee best results:
All variants in the VCF file which do not fulfil the following quality criteria will be filtered out by AION to avoid evaluating false positive variants. This means that even if your file has the required format and fields, it may contain low quality data, and wrong or no results would be produced.
Small variants:
CNV / SV variants:
AION CNV analysis does not hard filter on other quality metrics due the high variability of the scales of these metrics between different variant callers. However, AION CNV analysis considers as high quality and ranks higher variant fulfilling the criteria outlined above (VCF format | CNV / SV variants VCFs ).
In principle all variant callers can be used to generate the VCF for small variants, but some tools do not normalise variant calls by default (e.g. contain multi-allelic variants that have not been standardised following best practices). This can result in variant annotation problems: please revise that variant calls have been normalised before submitting the files.
For CNVs and SVs, AION has been validated on Canvas, Manta, Dragen CNV and Dragen SV. Other variant callers may be supported if they adhere to the same standards.
In case there is an error, the user is informed through the following messages:
| Error code | Category | Explanation | What to do |
| E103 | General | Failed on decompressing input file. Only gzipped and bgzipped vcf files are supported | Use another tool to compress the VCF file or contact support if needed |
| E104 | General | Some of the provided gene symbols or HGNC IDs are invalid | Validate the input and contact support if needed |
| E140 | VCF integrity | Provided samples do not have unique sample IDs. There are more than one files with the same sample ID | Merge the VCFs with repeated IDs and submit again |
| E141 | VCF integrity | Required fields check failed for input file! There are some missing fields in the VCF that are required | See required fields above and contact support if needed |
| E142 | VCF integrity | Import check failed for input file | Contact support |
| E143 | VCF integrity | VCF file could not be checked for mandatory columns | Contact support |
| E144 | VCF integrity | VCF file is missing the some columns | See required fields above and contact support if needed |
| E145 | VCF integrity | VCF file truncated | Validate the input and contact support if needed |
| E146 | VCF integrity | GVCF file detected. | GVCF is currently not a supported input format, please convert to VCF. Contact support if needed |
| E147 | VCF integrity | Structural variant file submitted to SNV pipeline or the other way around. | See information for how to submit a case Creating a case , contact support if needed. |
| E150 | VCF Processing | VCF consistency checks failed due to missing definitions in header | See required fields above and contact support if needed |
| E151 | VCF Processing | No variants in the selected region. Update your in silico panel filters if they were applied | If you added in silico panel filters, resubmit the case selecting other genes, otherwise the content of you VCF may not overlap with AION’s supported region. Contact support if needed. |
| E162 | Multisample VCF | Found more than one sample in input VCF! Currently, AION only supports singletons or trios. | Split your VCF into individual samples and submit again. Contact support if needed. |
| E170 | Reference genome mismatch | A mismatch was found between the reference genome provided and the input VCF files | Verify the provided reference genome is correct |
| E301 | Sample matching | The sample ID in the small and structural variants VCFs doesn’t match for the same samples. | Verify the sample IDs in the VCF files |
Guidelines for VCF files: https://samtools.github.io/hts-specs/VCFv4.3.pdf
Tools to validate VCF files:

